The normal QTc inteval of healthy individuals ranges between 350 to 450 ms for males and 360 to 460 ms for females. The short QT syndrome (SQTS) is a rare inherited primary electric cardiac disease characterized by abnormally short QT intervals on the ECG QTc ≤ 330 ms or QTc interval <360 ms and one or more of the following: history of cardiac arrest or syncope, family history of sudden cardiac death (SCD) at age 40 or younger or a family history of SQTS (Priori et al., 2013). The SQTs is associated with atrial and ventricular tachyarrhythmias, particularly at rest, and SCD (Bjerregaard et al., 2005; Patel et al., 2010). However, controversy exists about the exact cut-off value for the short QTc interval and because only a limited number of SQTS patients have been reported the prevalence of the syndrome is uncertain, but it seems that the occurrence of SCD as first manifestation is not infrequent.
Data from over 10,000 adults suggest that, in the healthy population, the prevalence of QTc <340 ms is approximately 0.5% (with 95 % confidence interval) (Anttonen et al., 2007). Therefore, males with QTc ≤330 ms and females with QTc ≤340 ms have abnormally short QT and should be considered to have SQTS, even if they are asymptomatic. However, individuals with QTc <320 ms who reached adulthood without life-threatening arrhythmias have been reported (Kobza et al., 2009).
1. Clinical presentation
The disease appears to be highly lethal in all age groups, including children in their first months of life, and the probability of a first cardiac arrest by the age of 40 years is >40%.One-third of the cases presented with sudden cardiac death as their first clinical manifestation, and 80 % of subjects were reported to have a personal or family history of SCD (Ferrero-Miliani et al., 2010). Some patients remain asymptomatic and are diagnosed due to strong family history, while other patients present palpitations, paroxysmal or permanent AF, syncope, ventricular arrhythmias and SCD. However, syncope is less frequent than in other arrhythmogenic syndromes. The onset of symptoms is highly variable, and episodes of SCD have been reported during or following loud noise, at rest, during exercise, and during daily activities. SCD has been described in the first year of life, suggesting that the SQTS can be the cause of death in some infants.
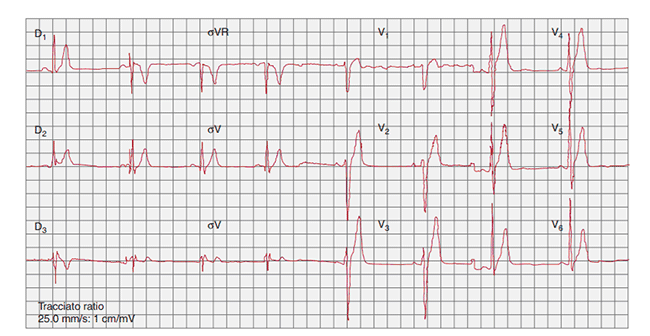
Figure. ECG of a patient with SQTS (QT/QTc, 300/300ms). Taken from Monforte et al., 2012
It is relatively simple and can be done only with the ECG and the patient's medical history (Table) (Gollob et al., 2011). SQTS should be strongly suspected in young individuals with a short QT interval on 12-lead ECG with arrhythmic symptoms, unexplained syncope or atrial fibrillation (AF) at a young age, resuscitated VF, and a strong family history of arrhythmic events including SCD (Patel et al., 2010). The QT interval should be measured preferable when the heart rate is <80 bpm, because all QTc formulae will overcorrect the true QTc intervals at higher heart rates, leading to a false negative diagnosis.
However, it is possible that a routine ECG does not show any abnormality and that the syndrome can be diagnosed in a later study or in a study of 24-48 hour Holter monitoring. An electrophysiology study helps to determine the ventricular refractory period and to evaluate the inducibility of VF and/or AF and can confirm the diagnosis. However, the presence of a short QT interval in the absence of symptoms or family history can not be considered as a SQTS.
Table. Diagnosis of SQTS diagnostic criteria (taken from Priori et al., 2015)
Recomendations
Class, Level
SQTS is diagnosed in the presence of a mQTc ≤340 ms.
I, C
SQTS should be considered in the presence of a QTc =360 ms and one or
more of the following:
(a) A confirmed pathogenic mutation
(b) A family history of SQTS
(c) A family history of sudden death at age ,40 years
(d) Survival from a VT/VF episosode
The ECG in SQT is characterized by abnormally short QT intervals, commonly ranging between 220 to 360 ms in patients with a heart rates between 60 y 85 bpm. QTc shortening can be intermittent and less evident in patients with SQT3 (~360 ms). T here is also a loss of QTc adaptation to changes in heart rate and the QT-RR relationship is less linear and its slope is less steep in the SQTS patient as compared with control subjects ( Patel et al., 2010 ). Patients with SQTS present ta ll, peaked and symmetrical T waves in the precordial leads , the ST segment is short or almost absent and the T wave originates from the S wave. In patients with SQT 3 , T waves are asymmetrical, with a less steep ascending limb followed by a rapid descending limb. In many ECG there is a p rolonged Tpeak-Tend interval and Tpeak-Tend/QT ratio, an indication of augmented transmural dispersion of repolarization (Gupta et al., 2008 ).
Secondary causes of short QT interval (i.e., hyperkalemia, acidosis, hypercalcemia, hyperthermia, digoxin, increased parasympathetic tone or increased plasma levels of catecholamines or testosterone) must be ruled out before considering the diagnosis of SQTS. A paradoxical ECG shortening of QT interval associated with a decrease in heart rate should also be considered in a differential diagnosis.
SQTS is a genetically heterogeneous autosomal dominant disease, but only 20% of symptomatic patients are successfully genotyped. To date, six genes encoding K+ and Ca2+ channels have been linked to the clinical manifestation of the disease. Therefore, the value of genetic testing is limited and does not bear prognostic implications. Thus, further studies are needed to identify new genes and to establish a genotype-phenotype correlation.
Mutations in 6 different genes (Table) can result in a QT interval phenotype opposite to that of LQTS. SQTS is associated with gain-of-function mutations in genes encoding outward K+ channels (KCNH2, KCNQ1, and KCNJ2) and a loss-of-function mutations in genes (CACNA1C, CACNB2B and CACNA2D1) encoding different subunits of cardiac L-type Ca2+ channel (Gussak et al., 2000; Gaita et al., 2003; Bellocq y cols., 2004; Brugada et al., 2004; Priori et al., 2005; Antzelevitch et al., 2007). A reduction in inward repolarizing currents and/or an increase in outward repolarizing currents will favor early repolarization, leading to a shortening of atrial and ventricular APD (QT interval) and transmural dispersion of repolarization and provides an ideal substrate for the development of reentrant mechanisms, which in the atria can lead to AF and in the ventricles to VF ant ventricular arrhythmias (Antzelevitch C, et al., 2007; Bjerregaard et al., 2005; Templin et al., 2011).
Invasive electrophysiologic studies revealed that atrial ERPs are extremely short in SQTS (120–180 ms) and atrial vulnerability also is increased, with AF often being inducible by atrial programmed stimulation. Using an experimental model of SQT1 in which the genetically mediated gain of function of IKr was pharmacologically mimicked using the IKr agonist PD118057, Nof et al (2010) demonstrated the development of a prominent dispersion of repolarization and refractoriness between the crista terminalis and pectinate muscle in the canine right atrium. A single premature beat introduced during this vulnerable window readily induced AF. The reentrant substrate was generated by an abbreviation of the ERP and amplified by the spatial dispersion of repolarization within the atria. Quinidine, but not lidocaine, was effective in preventing AF in this setting.
A missense mutation (V141M) in KCNQ1 was reported in a neonate presenting with in utero bradycardia and postpartum short QT intervals and AF (Hong et al., 2005). Computer modeling showed that the mutation would shorten action potential duration of human ventricular myocytes and abolish the pacemaker activity of the sinoatrial node.
Mutations in K+ channels lead to increase in outward current during the plateau phase of the AP which accelerates ventricular repolarization and shortens the QT interval. The gain-of-function mutation KCNH2 N588K located at the S5-P loop region at the outer mouth of the channel leads to a loss of rectification of the IKr at physiological range of plateau voltages and shifts the voltage-dependent inactivation by +90 mV, resulting in a large IKr during the plateau phase of the AP and marked abbreviation of the AP and QT interval (Brugada et al., 2004). Interestingly, this mutation reduces the affinity of the IKr blockers, which has direct implication on the treatment of SQT1. The KCNQ1 V307L mutation causes a shift of -20 mV in the voltage dependence of activation of IKs and accelerates the activation kinetics resulting in gain-in-function (Bellocq et al., 2004). Interestingly, this mutation has also been described in a patient with AF (Chen et al., 2003). A gain-of-function mutation mutation (D172N) in the KCNJ2gene encoding for the strong inwardly rectifying channel protein Kir2.1 increases the IK1 at potentials between -75 and -45 mV and the peak current is shifted from -75 mV to – 65 mV (Priori et al., 2005). As a result ventricular repolarization is greatly accelerated and the APD shortened.
A reduction in depolarizing currents can also shorten the QT interval. Loss-of-function mutations in the CACNA2D1 gene (Ser755Thr), encoding for the auxiliary Cavα2δ-1 subunit involved in the forward trafficking, membrane turnover, and modulation of biophysical properties of the pore-
forming a1 subunits has been described in patients with SQT6, leading to a decreased calcium current (Temlin et al., 2011). Mutations in CACNB2b (S481L) and CACNA1C (A39V and G490R) also cause a major loss of function in calcium channel activity (Antzelevitch et al., 2007).
Table. Genetic loci and genes associated with the Short QT Syndrome
The optimal strategy for primary prevention of cardiac arrest in SQTS is unclear, given the lack of independent risk factors for cardiac arrest, including syncope (Mazzanti et al. 2014). No data are available to quantify the risk of arrhythmic events during competitive physical activity in SQTS patients.
At present, there is no pharmacological therapy to prevent life-threatening arrhythmias, and because the rate of recurrence of cardiac arrest has been estimated at 10% per year, the ICD is is recommended in patients with a diagnosis of SQTS who (a) Are survivors of an aborted cardiac arrest, and/or (b) have documented spontaneous sustained VT with or without syncope (Recommendation I, C) (Priori et al., 2015). The decision to insert ICD should be based on clinical grounds (short QT interval on the ECG together with arrhythmic symptoms and strong family history of SCD) rather than genetic or electrophysiological until further guidelines for risk stratification are available (Patel et al., 2010). The main problem is that the tall and peaked T wave that closely follows the R wave can sometimes be interpreted as a short R–R interval, provoking an inappropriate shock from the ICD (Schrimpf et al., 2003).
Pharmacological therapy may be useful as an adjunct to the ICD or may be used for primary prevention when the patient refuses an ICD or in young children in whom the implantation of an ICD may be problematic. Some patients respond to the treatment with quinidine (or hydroxyquinidine) which inhibits the Ito, normalizes the QT interval and decreases the amplitude of the T wave of the ECG. Preliminary data suggest a benefit of disopyramide in patients with SQT1, consistent with the experimental data in N588K KCNH2 mutant channels (Schimpf et al., 2007; Gaita et al., 2004). However, flecainide, ibutilide and sotalol do not modify the QT interval. Flecainide prolongs the refractory period, but not the induction of VF in programmed stimulation. Most QT-prolonging drugs have the highest affinity to the inactivated state of IKr, but the mutations in KCNH2 responsible for SQTS1 impair inactivation of IKr, which explains the relative resistance to these drugs (Gaita et al., 2004; Perrin et al., 2008). Quinidine has similar affinity to the open and inactivated states of IKr and in small cohorts of patients quinidine therapy can prolong the QTc interval and possibly reduce arrhythmic events (Gaita et al., 2004; Giustetto et al., 2011). Patients on quinidine should be carefully monitored for QT prolongation and possible pro-arrhythmic events. Quinidine or sotalol may be considered in patients with a diagnosis of SQTS who qualify for an ICD but present a contra-indication to the ICD or refuse it (Recommendation IIb, C); quinidine or sotalol may be considered in asymptomatic patients with a diagnosis of SQTS and a family history of SCD (Recommendation IIb, C) (Priori et al., 2015).
AF is a common clinical problem in SQTS. Propafenone has been shown to be effective in preventing frequent paroxysms of AF with no recurrence of arrhythmia for more than two years without any effect on QT interval (Bjerregaard and Gussak, 2005).
An ICD might be considered on a case-by-case basis in patients with SQTS with a strong family history of SCD and evidence for abbreviated QTc in at least some of the patients, but there are not enough data to make generalized recommendations (Priori et al., 2013).
Invasive EPS with programmed ventricular stimulation is not recommended for SCD risk stratification (Priori et al., 2015).
Anttonen O, Junttila MJ, Rissanen H, et al. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation 2007;116:714–20.
Antzelevitch C, Pollevick GD, Cordeiro JM et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–9.
Bjerregaard P, Gussak I. Short QT syndrome: mechanisms, diagnosis and treatment. Nat Clin Pract Cardiovasc Med. 2005;2:84–87.
Bjerregaard P, Gussak I. Short QT syndrome. Ann Noninvasive Electrocardiol 2005;10:436–40.
Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004;109:2394-7.
Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004; 109: 30–35.
Chen YH, Xu SJ, Bendahhou S, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–254.
Ferrero-Miliani L, Holst AG, Pehrson S, et al. Strategy for clinical evaluation and screening of sudden cardiac death relatives. Fundam Clin Pharmacol. 2010;24:619–35.
Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: a familial cause of sudden death. Circulation 2003;108:965–70.
Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment, J Am Coll Cardiol 2004;43:1494–9.
Giustetto C, Schimpf R, Mazzanti A, et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol 2011;58:587–595.
Gupta P, Patel C, Patel H, et al. Tp-e/QT ratio as an index of arrhythmogenesis. J Electrocardiol 2008;41:567–574.
Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome?. Cardiology. 2000;94:99-102.
Hong K, Piper DR, Diaz-Valdecantos A, et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero, Cardiovasc Res 2005;68:433–40.
Kobza R, Roos M, Niggli B, et al. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm 2009;6:652–7.
Mazzanti A, Kanthan A, Monteforte N, et al. Novel insight into the natural history of short QT syndrome. J Am Coll Cardiol 2014;63:1300–1308.
Monteforte N, Napolitano C, Napolitano C, et al. Genetics and arrhythmias: diagnostic and prognostic applications. Rev Esp Cardiol 2012;65:278-285.
Nof E, Burashnikov A, Antzelevitch C. Cellular basis for atrial fibrillation in an experimental model of short QT1: implications for a pharmacological approach to therapy. Heart Rhythm. 2010;7:251-7.
Patel C, Yan G-Z, Antzelevitch C. Short QT Syndrome: From bench to bedside. Circ Arrhythm Electrophysiol. 2010;3:401-408.
Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005; 96: 800–807.
Priori SG, Wilde AA, Horie M, et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm 2013;10:e85–e108.
Priori SG, Blomströ m-Lundqvist C, Mazzanti A, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J 2015;36:2793–2867.
Schimpf R, Antzelevitch C, Hsu LF, et al. The QT-interval in patients with a Brugada syndrome: is a shortening of the QT-time an existing and relevant ECG-pattern? Heart Rhythm. 2007;4:S188.
Schimpf R, Veltmann C, Giustetto C, et al. In vivo effects of mutant HERG K+ channel inhibition by disopyramide in patients with a short QT-1 syndrome: a pilot study. J Cardiovasc Electrophysiol 2007;18:1157–60.
Schimpf R, Wolpert C, Bianchi F, et al. Congenital short QT syndrome and implantable cardioverter defibrillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol, 2003;14:1273–7.
Templin C, Ghadri JR, Rougier JS et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6) Eur Heart J. 2011;32:1077–88.