|
Prevalence The BrS is a channelopathy transmitted as an autosomal dominant trait with incomplete penetrante and male predominance and an abnormal ECG pattern characterized by ST-segment elevation (= 2 mm) of the right precordial leads (V1-V3), with or without right bundle branch block in patients with structurally normal hearts (Brugada et al., 1992; Antzelevitch et al., 2005). Patients with BrS present a high incidence of syncope and primary VF often resulting in SCD, which can be the first clinical manifestation (Brugada 1992, 2002, 2003; Mizusawa et al., 2012). The prevalence of BSr is estimated 1-5/10.000 inhabitants, but it can be much higher, since many patients are asymptomatic (Mizusawa and Wilde, 2012). The prevalence of BrS with a type 1 ECG in adults is higher in Asian countries, such as Japan (0.15–0.27%) and the Philippines (0.18%), and among Japanese-Americans in North America (0.15%) than in western countries, including Europe (0%–0.017%)and North America (0.005–0.1%) (Antzelevitch et al., 2016). In fact, in young men with normal hearts of some Southeast Asian countries the BrS is the second cause of death, surpassed only by car accidents (Veerakul and Nademanee, 2012). We don´t know how gender modulates the manifestation of the disease (Wilde et al., 2002; Antzelevitch et al., 2005). In pre-pubescent individuals, there are no significant gender differences in all 3 ST levels (ST-J, -M, and -E) in both leads V2 and V5, but levels increase significantly after puberty in males (Ezaki et al., 2010). Androgen-deprivation therapy significantly lowered all 3 ST levels in both V2 and V5 and closely resembled the ST levels in age-matched control females, suggesting that testosterone modulates the ion currents underlying the early phase of ventricular epicardial repolarization (AP notch). Furthermore, the effect of estrogens (they reduce Ito density and protein expression of the underlying Kv4.3 channels) and the differences in expression and density of the Ito current between both sexes (Ito density in the epicardium is higher in males than females) can also explain the predominance of BS phenotype among males)(Di Diego et al., 2002). The BrS is attributed to mutations on different genes leading to a shortening of the cardiac AP due to a decrease in inward-depolarizing currents (INa and ICa) or an increase in repolarizing currents (Ito or IKATP) (Table). However, genetic heterogeneity of BrS is likely to be even geater as mutation screening on the known genes allows identifying a mutation in ~25-30% of clinically affected patients. As a consequence, the value of genetic testing for diagnostic purposes is limited and there is no evidence that results of genetic testing influence clinical management or risk stratification in BrS (Priori et al., 2012). Because of the low prevalence of non- SCN5A mutations, it has been suggested that it is reasonable to initially test most patients for SCN5A mutations alone, with further testing for the other minor BS genes only in special circumstances (Ackerman et al., 2011). 1. Romano-Ward syndrome. At present, 15 different variants of the Romano-Ward syndrome have been published resulting from mutations in genes coding for cardiac proteins including ion channels, accessory subunits, and associated modulator proteins, responsible for orchestrating the cardiac AP. The first three LQTS genes identified were: KCNQ1 (LQT1), KCNH2 (LQT2), and SCN5A (LQT3) encoding for the proteins that conducts IKs, IKr and INa, respectively. Mutations in KCNQ1 and KCNH2 cause a decrease in the corresponding K+ current, while mutations in the SCN5A gene cause a gain-of-function phenotype leading to a prolongation of the ventricular AP repolarization and of the QTc interval (Curran et al., 1995; Goldenberg and Moss, 2008). Mutations in the 3 major LQTS-susceptibility genes (KCNQ1, KCNH2 and SCN5A) account for approximately 60–75% of congenital LQTS cases with a strong clinical phenotype, while the 15 other minor genes contribute approximately an additional 5% (Tester et al., 2006). The frequency of clinical events prior to initiation of beta-blocker therapy from birth to 40 years of age is significantly higher in LQT2 (46%) and LQT3 (42%) patients relative to those with LQT1 (30%) (Priori et al., 2003), but events in LQT3 are more likely to be lethal (Zareba et al., 1998).
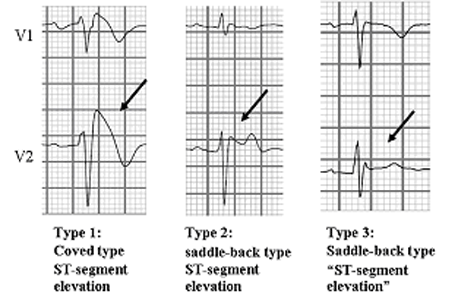
The electrophysiological background of arrhythmias in BrS is not fully understood. There is a debate on whether the BrS is a repolarization disorder [namely, transmural dispersion of right ventricular (RV) AP morphology, driven by the loss of the spike and dome action potential morphology at RV epicardium] or a depolarization disorder, namely RV conduction delay. A current hypothesis is that the BrS phenotype occurs when there is an imbalance between outward and inward currents at the end of phase 1 of the epicardial ventricular AP (Di Diego et al., 2002). Epicardial cells display a characteristic spike-and-dome morphology due to large transient outward K+ current (Ito) and short APD resulting from a high density of IKs, while Ito density is less marked in the endocardial cells (Yan and Antzelevitch, 1999; Antzelevitch, 2005). Mutations reducing INa or I Ca amplitude in the presence of large repolarizing currents (Ito and IKs) may result in a rapid repolarization phase 1, disappearance of the dome and a marked shortening of the AP in the epicardial, but not in the endocadial cells. This creates a transmural voltage gradient that may be responsible the characteristic ST-segment elevation and a favorable substrate for ventricular arrhythmias due to a mechanism of phase 2 reentry. The RV outflow tract epicardium (RVOT) has a higher Ito density compared with the LV, which explains why only the right precordial leads present the coved-type ST-segment elevation. Reduced myocardial Na+ current (or Na+ channel blockers) will cause disproportionate shortening of epicardial AP because of unopposed Ito, lead to an exagerated transmural voltage gradient, increase ST segment elevation and unmask a concealed the type 1 ECG pattern. However, it is difficult to understand why mutations affect almost selectively the RV (not the entire heart) or why the type 1 ECG pattern is intermittently present. Thus, it is likely that the presence of a mutation is required but not sufficient to produce the electrical 'signature' of the disease. If so, the presence of structural abnormalities may play an important role (Frustaci et al., 2005; Yan et al., 1999). The depolarization hypothesis suggests that slow conduction in the RVOT, secondary to fibrosis and reduced Cx43 leading to discontinuities in indeterminate conduction, plays a primary role in the development of the ECG and arrhythmic manifestations of the BrS (Nagase et al., 2002, Postema et al 2008; Wilde et al., 2010; Elizari et al., 2007). Conduction slowing is not necessarily limited to the RVOT area. It has been proposed that changes in ion channel current responsible for BrS (i. e., loss of function INa and ICa and gain of function of Ito) can alter AP morphology so as to reduce the safety of conduction at high resistance junctions, such as regions of extensive fibrosis (Hoogendijk et al. 2010). However, the typical behavior of patients with BrS to acceleration of rate is diminution of ST-segment elevation, opposite to that expected at a site of discontinuous conduction. It has been demonstrated significant regional conduction delay, reduction in activation gradient and formation of lines of functional conduction block in the anterolateral free wall of the right ventricular outflow tract compared with the right ventricular body and apex of BS patients (Lambiase et al., 2009). Moreover, fractionated electrograms in the RV, possibly due to subtle structural abnormalities (hypertrophy, vacuolation and cardiomyocyte apoptosis, fibrofatty infiltration), have been also reported in patients with BS (Coronel et al., 2005; Postema et al., 2010). The delay in the AP of the RVOT causes an electrical gradient from the more positive RV to the RVOT, leading to ST-elevation in the right precordial leads and as the RVOT depolarizes later (during repolarization of the RV) this gradient is reversed and the net current flows towards the RV, resulting in a negative T-wave in the same right precordial leads (Meregalli et al., 2005). More recently, Nademanedee et al (2011) found in patients with a type 1 pattern BrS and episodes of VT/VF abnormal low voltage, prolonged duration, and fractionated late potentials clustering exclusively in the anterior aspect of the right ventricular outflow tract epicardium. These results confirm the presence of delayed depolarization at this site that would facilitate the development of epicardial reentry circuits and would be aggravated by the reduction of the INa. Under these circumstances, Na+ channel blockers create additional conduction delay between the other part of the ventricles and the RVOT (the depolarization theory) or induce the transmural gradient of AP by shortening AP more in the RVOT epicardium than endocardium (the repolarization theory), which, theoretically, both lead to the manifestation of coved-type ECG (Meregalli et al., 2005). There is another explanation, i.e. the “developmental” hypothesis, in which abnormal expression of cardiac neural crest cells in the ROVT leads to abnormal connexin expression (Cx43) and combined depolarization–repolarization abnormalities favor¬ing arrhythmia (Elizari et al., 2007). They are quite variable, from asymptomatic patients to those in which the first manifestation is a SCD (Antzelevitch et al., 2005). The most common clinical manifestations are syncope or seizures, agonal respiration or SCD caused by self-terminating VF episodes mostly occurring during sleep or at rest. Sinus function is normal, although supraventricular tachycardias, the most frequent atrial fibrillation (AF), are present in 20-30% of patients; indeed, AF can be the first manifestation of the BrS (Kusano et al., 2008). Atrioventricular block and intraventricular conduction delays (HV interval of 60-75 ms) are also part of the phenotype of BrS (Smits et al., 2002), and a high percentage of patients have inducible VT (or VF) during programmed ventricular stimulation. The intraventricular conduction delays explain the slight prolongation of the PR interval and the morphology of right bundle branch block and left anterior hemiblock in the ECG. Interestingly, hemodynamic studies are normal. The ECG pattern ECG morphology is highly variable over time even in the same patient, so that in asymptomatic individuals the typical ECG syndrome can be found by chance during a routine examination or during a study because of a family history of SCD. Conversely, in some patients the diagnosis is reached because of unexplained or vasovagal syncope or idiopathic VF, or because they were challenged with class I antiarrhythmic drugs that unmask a concealed or non-diagnostic ECG pattern (Figure 1). T hree patterns have been described (Antzelevitch et al., 2005)(Figure 1). It is possible that the electrocardiographic patterns differ depending on the mutation and that the observed variations over time are related to changes in autonomic tone, body temperature or heart rate (Benito et al., 2008; Mizusawa et al., 2012). Adrenergic stimulation (isoproterenol), exercise and an increase in heart rate decrease the ST segment elevation; indeed, some patients with “VF storms” associated with BrS can be effectively treated with isoproterenol infusion (Tanaka et al., 2001).
According to the 2013 consensus statement on inherited cardiac arrhythmias (Priori et al., 2013) and the 2015 guidelines for the management of patients with ventricular arrhythmias and prevention of SCD (Priori et al., 2015): "BrS is diagnosed in patients with ST- segment elevation with type 1 morphology =2 mm in =1 lead among the right precordial leads V1, V2, positioned in the 2nd, 3rd or 4th intercostal space occurring either spontaneously or after provocative drug test with intravenous administration of Class I antiarrhythmic drugs. BrS is diagnosed in patients with type 2 or type 3 ST-segment elevation in =1 lead among the right precordial leads V1, V2 positioned in the 2nd, 3rd or 4th intercostal space when a provocative drug test with intravenous administration of Class I antiarrhythmic drugs induces a type I ECG morphology.” In any case, it is necessary to rule out other conditions producing a ST segment elevation of the ECG (ischemia, myocarditis, hyperkalemia, hypercalcemia, ventricular arrhythmogenic dysplasia or pulmonary embolism).
Education and lifestyle changes for the prevention of arrhythmias are critical in BrS. All patients with a Brugada ECG should be taught to treat aggressively any episodes of fever and to avoid drugs known to exacerbate the condition (http://www.brugadadrugs.org). Ackerman M, Priori S, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Heart Rhythm 2011;8:1308-1339. Agac MT, Erkan H, Korkmaz L. Conversion of Brugada type I to type III and successful control of recurrent ventricular arrhythmia with cilostazol. Arch Cardiovasc Dis 2014;107:476–478. Aizawa Y, Yamakawa H, Takatsuki S, et al. Efficacy and safety of bepridil for prevention of ICD shocks in patients with Brugada syndrome and idiopathic ventricular fibrillation. Int J Cardiol 2013;168:5083–5. Akai J, Makita N, Sakurada H, Set al. A novel SCN5A mutation associated with idio¬pathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Lett. 2000;479:29–34. Amin A, Asghari A, Tan HL. Cardiac sodium channelopathies. Pflugers arch 2010;460:223-237. Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: Report of the Second Consensus Conference: Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659-670. Antzelevitch C, Fish JM. Therapy for the Brugada syndrome. Handb Exp Pharmacol 2006;171:305–330. Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007;115:442–449. Antzelevitch C. Role of transmural dispersion of repolarization in the genesis of drug-induced torsades de pointes. Heart Rhythm. 2005;2(2 Suppl):S9–S15. Antzelevitch C, Patocskai B. Brugada Syndrome: Clinical, Genetic, Molecular, Cellular, and Ionic Aspects. Curr Probl Cardiol. 2016;41:7-57. Antzelevitch C, Yan GX, Ackerman MJ, et al. J-Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. J Arrhythm. 2016;32:315-339. Barajas-Martinez H, Hu D, Ferrer T. Molecular genetic and functional association of Brugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm. 2012;9:548–555. Baroudi G, Pouliot V, Denjoy I, et al. Novel mechanism for Brugada syndrome: defective surface localization of an SCN5A mutant (R1432G). Circ Res 2001;88:E78-83. Behr ER, Savio-Galimberti E, Barc J, et al. Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc Res 2015;106:520–9. Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome.Circulation 2004;110:1731–1737. Belhassen B, Rahkovich M, Michowitz Y, et al. Management of Brugada syndrome: a 33-year experience using electrophysiologically-guided therapy with Class1A antiarrhythmic drugs. Circ Arrhythm Electro- physiol 2015;6:1393–402. Belhassen B, Viskin S, Antzelevitch C. The Brugada syndrome: is an implantable cardioverter defibrillator the only therapeutic option? Pacing Clin Electrophysiol 2002;25:1634–1640. Benito B, Sarkozy A, Mont L, Henkens S, Berruezo A, Tamborero D, et al. Gender differences in clinical manifestations of Brugada syndrome. J Am Coll Cardiol. 2008;52:1567-73. Berne P, Brugada J. Brugada syndrome 2012. Circ J 2012;76:1563-1571. Bezzina C, Veldkamp MW, van den Berg MP, et al. A single Na+ chan¬nel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–1213. Bezzina CR, Barc J, Mizusawa Y. Common variants at SCN5A-SCN10Aand HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013;45:1044–1049. Boczek NJ, Ye D,Johnson EK, et al. Characterization of SEMA3A-encoded semaphorin as a naturally occurring Kv4.3 protein inhibitor and its contribution to Brugada syndrome.CircRes2014;115:460–9. Bouzeman A, Traulle S, Messali A, et al. Long-term follow-up of asymptomatic Brugada patients with inducible ventricular fibrillation under hydroquinidine. Europace 2014;16:572–577. Brugada P, Brugada J, Roy D. Brugada syndrome 1992–2012: 20 years of scientific excitement, and more. Eur Heart J 2013;34:3610–3615. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391-1396. Brugada J, Brugada R, Antzelevitch C, et al. Long-term follow-up of individuals with the electrocardiographic pattern of right bundle-branch block and ST-segment elevation in precordial leads V1 to V3. Circulation. 2002;105:73-78. Brugada J, Brugada R, Brugada P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome and no previous cardiac arrest. Circulation. 2003;108:3092-3096. Brugada P, Brugada R, Brugada J. Patients with an asymptomatic Brugada electrocardiogram should undergo pharmacological and electrophysical testing. Circulation 2005;112:279–285. Brugada J, Brugada R, Brugada P. Pharmacological and device approach to therapy of inherited cardiac diseases associated with cardiac arrhythmias and sudden death. J Electrocardiol. 2000;33(Suppl):41–47. Brugada J, Pappone C, Berruezo A, et al. Brugada syndrome phenotype elimination by epicardial substrate ablation. Circ Arrhythm Electrophysiol. 2015;8:1373–1381. Burashnikov E, Pfeiffer R, Barajas-Martinez H, et al. Mutations in the cardiac L-type calcium channel associ¬ated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm. 2010;7:1872–1882. Cerrone M, Lin X, Zhang M, et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2014;129:1092–103. Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998;392:293–296. Chiale PA, Garro HA, Fernandez PA, et al. High-degree right bundle branch block obscuring the diagnosis of Brugada electrocardiographic pattern. Heart Rhythm 2012;9:974–6. Chinushi M, Aizawa Y, Ogawa Y, et al. Discrepant drug action of disopyramide on ECG abnormalities and induction of ventricular arrhythmias in a patient with Brugada syndrome. J Electrocardiol. 1997;30:133–136. Conte G, Sieira J, Ciconte G, et al. Implantable cardioverter-defibrillator therapy in Brugada syndrome: a 20-year single-center experience. J Am Coll Cardiol 2015;65:879–88. Coronel R, Casini S, Koopmann TT, et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: A combined electrophysiological, genetic, histopathologic, and computational study. Circulation 2005;112:2769-2777. Delpon E, Cordeiro JM, Nunez L, Thomsen PEB, Guerchicoff A, Pollevick GD, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1:209-18. Di Diego JM, Cordeiro JM, et al. Ionic and cellular basis for the predominance of the Brugada syndrome phenotype in males. Circulation 2002;106:2004–2011. DiFrancesco D. Funny channel gene mutations associated with arrhythmias. J Physiol. 2013;591:4117–4124. Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are tem¬perature dependent. Circ Res. 1999;85:803–809. Elizari MV, Levi R, Acunzo RS, et al. Abnormal expression of cardiac neural crest cells in heart development: a different hypothesis for the etiopathogenesis of Brugada syndrome. Heart Rhythm. 2007;4: 359–365. Ezaki K, Nakagawa M, Taniguchi Y, et al. Gender differences in the ST segment: Effect of androgendeprivation therapy and possible role of testosterone. Circ J 2010;74:2448-2454. Frustaci A, Priori SG, Pieroni M, et al. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 2005;112:3680-3687. Fukuyama M, Ohno S, Makiyama T, et al. Novel SCN10A variants associated with Brugada syndrome. Europace 2015;18:905–11. George AL. Inherited disorders of voltage-gated sodium channels. J. Clin. Invest.2005; 115:1990–1999. Giudicessi JR, Ye D, Tester DJ, et al. Transient outward current (Ito) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm 2011;8:1024-1032. Grant AO, Carboni MP, Neplioueva V, et al. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J Clin Invest 2002;110:1201–1209. Hermida JS, Denjoy I, Clerc J, et al. Hydroquinidine therapy in Brugada syndrome. J Am Coll Cardiol 2004;43:1853–1860. Hoogendijk MG, Opthof T, Postema PG, etal. The Brugada ECG pattern: a marker of channelopathy, structural heart disease, or neither?. Toward a unifying mechanism of the Brugada syndrome. Circ Arrhythm Electrophysiol 2010;3:283–90. Hu D, Barajas-Martinez H, Burashnikov E, et al. A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet. 2009;2:270-278. Hu D, Barajas-Martínez H, Medeiros-Domingo A, et al. A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Na(v)1.5 and K(v)4.3 channel currents. Heart Rhythm. 2012;9:760-766. Hu D, Barajas-Martinez H, Pfeiffer R, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol 2014;64:66–79. Huang J-M, Horie M. Genetics of Brugada syndrome. J Arrhythm. 2016; 32: 418–425. Ishikawa T, Sato A, Marcou CA, et al. A novel disease gene for Brugada syndrome: sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hNav1.5. Circ Arrhythm Electrophysiol 2012;5:1098–107. Juang JJ, Horie M. Genetics of Brugada syndrome. J Arrhythm. 2016;32:418-425. Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010;7:33–46. Kattygnarath D, Maugenre S, Neyroud N, et al. MOG1: a new susceptibility gene for Brugada syndrome. Circ Cardiovasc Genet. 2011;4:261-268. Kusano KF, Taniyama M, Nakamura K, et al. Atrial fibrillation in patients with Brugada syndrome relationships of gene mutation, electrophysiology, and clinical backgrounds. J Am Coll Cardiol. 2008;51:1169-75. Kyndt F, Probst V, Potet F, et al. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation. 2001;104:3081–3086. Lambiase PD, Ahmed AK, Ciaccio EJ, Brugada R, Lizotte E, Chaubey S, Ben-Simon R, Chow AW, Lowe MD, McKenna WJ. High-density substrate mapping in Brugada syndrome: combined role of conduction and repolarization heterogeneities in arrhythmogenesis. Circulation 2009; 120:106-117, 101-104. Liu H, Chatel S, Simard C, et al. Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PLoS One 2013;8:e54131. London B, Michale M, Mehdi H, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPDL-1) decreases cardiac Na-current and causes inherited arrhthmias. Circulation. 2007;116:2260-2268. Makita N, Behr E, Shimizu W, et al. The E1784K mutation in SCN5A is associated with mixed clinical phenotype of type 3 long QT syndrome. J Clin Invest. 2008;118:2219–2229. Maury P, Couderc P, Delay M, et al. Electrical storm in Brugada syndrome successfully treated using isoprenaline. Europace 2004;6:130–133. Maury P, Hocini M, Haissaguerre M. Electrical storms in Brugada syn¬drome: review of pharmacologic and ablative therapeutic options. Indian Pacing Electrophysiol J. 2005;5:25–34. Medeiros-Domingo A, Tan BH, Crotti L, et al. Gain-of-function mutation S422L in the KCNJ8-encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm. 2010;7:1466-1471. Meregalli PG, Tan HL, Probst V, et al. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm 2009;6:341-348. Meregalli PG, Wilde AA, Tan HL. Pathophysiological mechanisms of Brugada syndrome: depolarisation disorder, repolarisation disorder or more? Cardiovasc Res. 2005;67:367-378. Mizasuwa Y, Wilde AA. Brugada syndrome. Circ Arrhythm Electrophysiol 2012;5:606-616. Mizusawa Y, Sakurada H, Nishizaki M, et al. Effects of low-dose quinidine on ventricular tachyarrhythmias in patients with Brugada syndrome: low-dose quinidine therapy as an adjunctive treatment. J Cardiovasc Pharmacol. 2006;47:359–364. Mok NS, Chan NY, Chi-Suen CA. Successful use of quinidine in treatmentof electrical storm in Brugada syndrome. Pacing Clin Electrophysiol 2004;27: 821–823. Murakami M, Nakamura K, Kusano KF, et al. Efficacy oflow-dose bepridil for preventionofventricular fibrillation in patients with Brugada syndrome with and without SCN5A mutation. J Cardiovasc Pharmacol 2010;56:389–95. Nademanee K, Veerakul G, Chandanamattha P, et al. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation. 2011;123:1270–1279. Nagase S, Kusano KF, Morita H, et al. Epicardial electrogram of the right ventricular outflow tract in patients with the Brugada syndrome: using the epicardial lead. J Am Coll Cardiol 2002;39:1992-1995. Nielsen MW, Holst AG, Olesen SP, et al. The genetic component of Brugada syndrome. Front Physiol. 2013;4:179. Ohgo T, Okamura H, Noda T, et al. Acute and chronic management in patients with Brugada syndrome associated with electrical storm of ventricular fibrillation. Heart Rhythm 2007;4: 695-700. Ohno S, Zankov DP, Ding WG, et al. KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ Arrhythm Electrophysiol. 2011;4:352-361. Pellegrino PL, Di BM, Brunetti ND. Quinidine for the management of electrical storm in an old patient with Brugada syndrome and syncope. Acta Cardiol 2013;68:201–3. Postema PG, van Dessel PF, de Bakker JM, et al. Slow and discontinuous conduction conspire in Brugada syndrome: a right ventricular mapping and stimulation study. Circ Arrhythm Electrophysiol 2008;1:379-386. Postema PG, van Dessel PF, Kors JA, et al. Local depolarization abnormalities are the dominant pathophysiologic mechanism for type 1 electrocardiogram in brugada syndrome a study of electrocardiograms, vectorcardiograms, and body surface potential maps during ajmaline provocation. J Am Coll Cardiol 2010;55:789-797. Priori S.G., Blomstrom-Lundqvist C., Mazzanti A, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36:2757–2759. Priori SG, Gasparini M, Napolitano C, et al. Risk stratification in Brugada syndrome: results of the PRELUDE (PRogrammed ELectrical stimUlation preDictive valuE) registry. J Am Coll Cardiol 2012;59:37-45. Priori S.G., Wilde A.A., Horie M. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10:1932–1963. Priori SG, Napolitano C, Gasparini M, et al. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation. 2002;105:1342-7. Probst V, Denjoy I, Meregalli PG et al. Clinical aspects and prognosis of Brugada syndrome in children. Circulation 2007;115:2042–2048. Probst V, Veltmann C, Eckardt L,etal. Long-term prognosis of patients diagnosed with Brugada syndrome: results from the FINGER Brugada Syndrome Registry. Circulation 2010;121:635–643. Probst V et al. The psychological impact of implantable cardioverter defibrillator implantation on Brugada syndrome patients. Europace. 2011;13:1034–9. Riuro H, Beltran-Alvarez P, Tarradas A, et al. A missense mutation in the sodium channel beta2 subunit reveals SCN2B as a new candidate gene for Brugada syndrome. Hum Mutat 2013;34:961–6. Sacher F, Jesel L, Jais P, et al. Insight into the mechanism of Brugada syndrome: epicardial substrate and modification during ajmaline testing. Heart Rhythm. 2014;11:732–734. Sacher F, Probst V, Maury P. Outcome after implantation of cardioverter- defibrillator in patients with Brugada syndrome: a multicenter study—part 2. Circulation. 2013;128:1739–1747. Schulze-Bahr E, Eckardt L, Breithardt G, et al. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Hum. Mutat. 2003:21:651–652. Shah AJ, Hocini M, Lamaison D. Regional substrate ablation abolishes Brugada syndrome. J Cardiovasc Electrophysiol. 2011;22:1290–1291. Shimizu W, Kamakura S .Catecholamines in children with congenital long QT syndrome and Brugada syndrome. J Electrocardiol 2001;34:173–5. Shinohara T, Ebata Y, Ayabe R, et al. Combination therapy of cilostazol and bepridil suppresses recurrent ventricular fibrillation related to J-wave syndromes. Heart Rhythm 2014;11:1441–1445. Shirai N, Makita N, Sasaki K, et al. A mutant cardiac sodium channel with multiple biophysical defects associated with overlapping clinical fea¬tures of Brugada syndrome and cardiac conduction disease. Cardiovasc Res. 2002;53:348–354. Smits JP, Eckardt L, Probst V, et al. Genotype-phenotype relationship in Brugada syndrome: electrocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients. J Am Coll Cardiol 2002;40:350–356. Smits JP, Koopmann TT, Wilders R, et al. A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. J Mol Cell Cardiol 2005;38:969-981. Sroubek J, Probst V, Mazzanti A. Programmed Ventricular Stimulation for Risk Stratification in the Brugada Syndrome: A Pooled Analysis. Circulation. 2016;133:622-30. Sugao M, Fujiki A, Nishida K, et al. Repolarization dynamics in patients with idiopathic ventricular fibrillation: pharmacological therapy with bepridil and disopyramide. J Cardiovasc Pharmacol 2005;45:545–549. Sumi S, Maruyama S, Shiga Y, et al. High efficacy of disopyramide in the management of ventricular fibrillation storms in a patient with Brugada syndrome. Pacing Clin Electrophysiol. 2010;33:e53-6. Suzuki H, Torigoe K, Numata O, et al. Infant case with a malignant form of Brugada syndrome. J Cardiovasc Electrophysiol 2000;11:1277–1280. Takehara N, Makita N, Kawabe J, et al. A cardiac sodium channel mutation identified in Brugada syndrome associated with atrial standstill. J Intern Med. 2004;255:137-142. Tanaka H, Kinoshita O, Uchikawa S, et al. Successful prevention of recurrent ventricular fibrillation by intravenous isoproterenol in a patient with Brugada syndrome. Pacing Clin Electrophysiol 2001;24:1293–4. Tanaka H, Kinoshita O, Uchikawa S, et al. Successful prevention of recurrent ventricular fibrillation by intravenous isoproterenol in a patient with Brugada syndrome. Pacing Clin Electrophysiol 2001;1293-1294. Tester DJ, Will ML, Haglund CM, et al. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2005;2:507–517. Tsuchiya T, Ashikaga K, Honda T, et al. Prevention of ventricular fibrillation by cilostazol, an oral phosphodiesterase inhibitor, in a patient with Brugada syndrome. J Cardiovasc Electrophysiol 2002;13:698–701. Tsuchiya T, Ashikaga K, Honda T, et al. Ueda K, Hirano Y, Higashiuesato Y, et al. Role of HCN4 channel in preventing ventricular arrhythmia. J Hum Genet. 2009;54:115-121. Ueda K, Nakamura K, Hayashi T, et al. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J Biol Chem 2004;279:27194-27198. Valdivia CR, Tester DJ, Rok BA, et al. A trafficking defective, Brugada syn¬drome-causing SCN5A mutation rescued by drugs. Cardiovasc Res. 2004;62:53–62. van Den Berg MP, Wilde AA, Viersma TJW. Possible bradycardic mode of death and successful pacemaker treatment in a large family with features of long QT syndrome type 3 and Brugada syndrome. J Cardiovasc Electrophysiol. 2001;12:630–636. Veerakul G, Nademanee K. Brugada syndrome: two decades of progress. Circ J 2012;76:2713-2722. Verkerk AO, Wilders R, Schulze-Bahr E, et al. Role of sequence variations in the human ether-ago-go-related gene (HERG, KCNH2) in the Brugada syndrome 1. Cardiovasc Res 2005;68:441-453. Viskin S, Wilde AA, Tan HL, et al. Empiric quinidine therapy for asymptomatic Brugada syndrome: time for a prospective registry. Heart Rhythm 2009;6:401–4. Wang DW, Makita N, Kitabatake A., et al. Enhanced Na+ channel intermediate inactivation in Brugada syndrome. Circ. Re 2000; 87:E37-E43. Wang Q, Ohno S, Ding WG. Gain-of-function KCNH2 mutations in patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2014;25(5):522–530. Watanabe A, Fukushima KK, Morita H, et al. Low-dose isoproterenol for repetitive ventricular arrhythmia in patients with Brugada syndrome. Eur Heart J 2006;27:1579–83. Watanabe H, Koopmann TT, Le Scouarnec S, et al. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Invest 2008;118: 2260-2268. Weiss R, Barmada MM, Nguyen T, et al. Clinical and molecular heterogeneity in the Brugada syndrome: a novel gene locus on chromosome 3. Circulation. 2002;105:707–713. Wilde AA, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation 2002;106:2514–2519. Wilde AA, Postema PG, Di Diego JM, et al. The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J Mol Cell Cardiol. 2010;49:543–553. Wilde AA, Viskin S. EP testing does not predict cardiac events in Brugada syndrome. Heart Rhythm 2011;8:1598-1600. Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100:1660-1666. Zhang J, Sacher F, Hoffmayer K et al. Cardiac electrophysiological substrate underlying the ECG phenotype and electrogram abnormalities in Brugada syndrome patients. Circulation 2015;131:1950–1959. Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation 2006;114:e385–484. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
| Aviso legal Esta obra está bajo una licencia de Creative Commons Reconocimiento-NoComercial-SinObraDerivada 4.0 Internacional |
