|
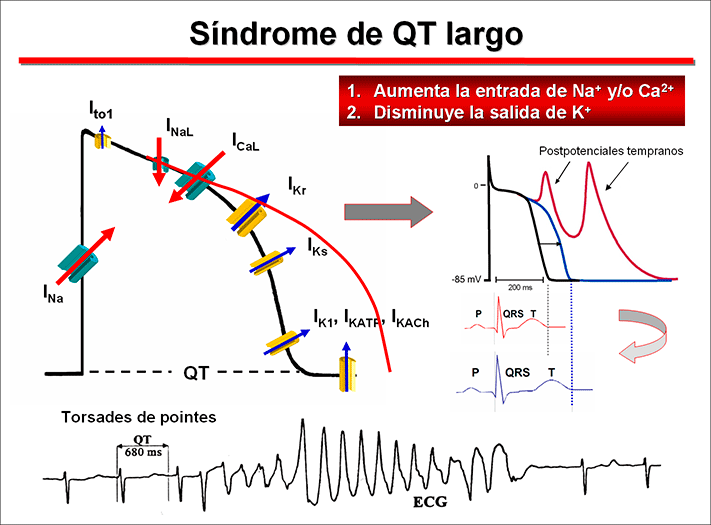
Prevalence Long QT syndrome (LQTS) is a group of disorders of cardiac repolarization characterized by an excessive and heterogeneous prolongation of the ventricular AP and of the corrected QT interval (QTc) of the surface (QTc Bazzet >440 ms in men, >460 ms in females) that predisposes affected subjects to syncope, seizures and polymorphic VT and/or VF and increases the risk of SCD (Goldenberg and Moss, 2008). It usually affects children and young adults apparently healthy, the average age of onset of symptoms (syncope or SCD) being 12 years of age or even earlier in the most serious forms of LQTS (Goldberg y Moss, 2008; Priori et al., 2003). Syncope and seizures associated with LQTS are caused by a specific polymorphic VT called torsade de pointes (“twisting of the points”) characterized by sinusoidal twisting of the QRS axis around the isoelectric line of an ECG tracing. Although the torsade de pointes are self-limited, sometimes they can degenerate into VF and SCD. The incidence of congenital LQTS has not been accurately defined, but it has been estimated that as many as 1 in 2,000-5,000 people worldwide are affected and a mortality rate of 21% for symptomatic patients not receiving therapy within one year from the first syncope event (Schwartz et al., 2012). However, given that up to 2/3 of patients are probably missed and that 10% to 35% have a normal corrected QT (QTc) interval it is likely that the actual prevalence is much higher. The Jervell-Lange Nielsen appears in 1.6-6 per million births (Schwartz et al., 2006). The main feature of the LQTS is the marked non-uniform prolongation of the ventricular APD and of the corrected QT interval (QTc) of the ECG resulting from an increase in the inward-depolarizing Na+ and Ca2+ currents and/or a decrease in outward-repolarizing K+ currents. The prolongation of the APD increases the dispersion in the recovery of ventricular excitability and facilitates the occurrence of early afterdepolarizations during the phase 2 and the beginning of phase 3. The early afterpotentials occurring during the proloned plateau of the AP would result from the activation of the INaL, the reactivation-reopening of the L-type Ca2+ channels or the activation of the current generated by the Na+-Ca2+ exchanger (INCX). The onset of early afterdepolarizations in the presence of an increased dispersion of refractoriness among the various ventricular regions can alter the pattern ventricular conduction, causing unidirectional block and slow conduction, conditions that may facilitate ventricular reentry.
1. Romano-Ward syndrome. At present, 15 different variants of the Romano-Ward syndrome have been published resulting from mutations in genes coding for cardiac proteins including ion channels, accessory subunits, and associated modulator proteins, responsible for orchestrating the cardiac AP. The first three LQTS genes identified were: KCNQ1 (LQT1), KCNH2 (LQT2), and SCN5A (LQT3) encoding for the proteins that conducts IKs, IKr and INa, respectively. Mutations in KCNQ1 and KCNH2 cause a decrease in the corresponding K+ current, while mutations in the SCN5A gene cause a gain-of-function phenotype leading to a prolongation of the ventricular AP repolarization and of the QTc interval (Curran et al., 1995; Goldenberg and Moss, 2008). Mutations in the 3 major LQTS-susceptibility genes (KCNQ1, KCNH2 and SCN5A) account for approximately 60–75% of congenital LQTS cases with a strong clinical phenotype, while the 15 other minor genes contribute approximately an additional 5% (Tester et al., 2006). The frequency of clinical events prior to initiation of beta-blocker therapy from birth to 40 years of age is significantly higher in LQT2 (46%) and LQT3 (42%) patients relative to those with LQT1 (30%) (Priori et al., 2003), but events in LQT3 are more likely to be lethal (Zareba et al., 1998).
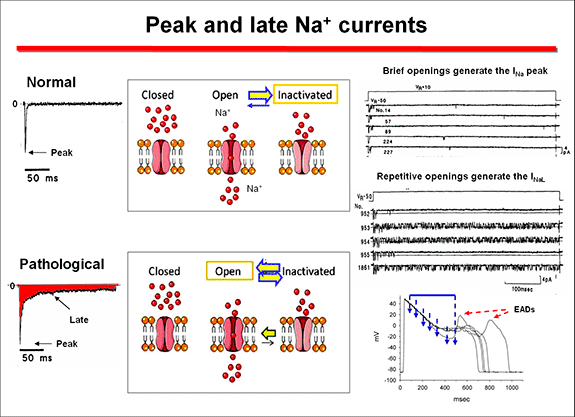
Na+ channel-linked mutations result in a gain-of-function from at least four distinct mechanisms. 1) Most of these mutations are missense mutations that disrupt the fast inactivation n, leading to bursting activity, which underlies the late Na+ current (INaL) over the plateau voltage range (Bennet et al., 1995). This increase in Na+ entry prolongs the plateau phase and produces a delayed repolarization that facilitates the induction of early afterdepolarizations that trigger the torsade de pointes (Yan et al., 2001). These mutations are localized in the a subunit regions implicated in the fast inactivation (i.e., S4 in DIV, DIII - DIV linker and cytoplasmic loops between S4 and S5 in DIII and DIV) or that stabilize the fast inactivation (C- terminal) (Tester et al., 2005, Zimmer and Surber, 2008). A general slowing of inactivation can be present in mutations associated with severe LQTS.
2) Less commonly the mutations facilitate the reopening of the channel (the so called"window current") over voltage ranges for which steady-state inactivation and activation overlap. The>INa increases when the inactivation of mutant channels occurs at more depolarized levels, while activation is unchanged, increasing the voltage range at which the channel can be reactivated and generate the INa (Wang et al., 1996). These two mechanisms exert their effects during phases 2 and 3 of the AP, where normally no (<1%) Na+ current is present. The delay in the repolarization process triggers early afterdepolarizations, especially in Purkinje and M cells, which present longer APD and induce torsades de pointes (Yan et al., 2001). 3) Mutations can induce a faster recovery from inactivation, leading to larger peak INa by increasing the fraction of channels available for activation during subsequent depolarizations (Amin et al., 2010). 4) Mutations can increase in the expresion of the a subunits in the cell membrane and, therefore, peak Na. This can result from an increased expression of mutant a subunits through enhanced mRNA translation or protein trafficking to the sarcolemma, decreased protein degradation, or altered modulation by β -subunits and regulatory proteins (Amin et al., 2010). Mutations occurring in the K+ channels can cause a loss of function and a decrease in outward K+ currents secondary because: Zhou et al (1998) described that some HERG mutations (Y611H and V822M) caused defects in biosynthetic processing of HERG channels with the protein retained in the endoplasmic reticulum (ER). ER-retained subunits are then rapidly degraded by the ubiquitin–proteasome pathway. Other mutations (I593R and G628S) were processed similarly to wild type HERG protein, but these mutations did not produce functional channels, while the T474I mutation expressed HERG current but with altered gating properties. At the present time, it is known that defective protein folding, retention in the ER, or disrupted trafficking to the Golgi and surface membrane is the primary mechanism of loss of function caused by missense mutations in HERG (Anderson et al., 2006). Mutations that enhance inactivation (e.g., G584S) (Zhao et al., 2009) or accelerate the rate of deactivation (e.g., M124R or other mutations in the PAS domain) (Sushi et al., 2005) reduce the IKr during repolarization of the action potential. LQT2 nonsense mutations cause a decrease in mutant mRNA levels by nonsense-mediated mRNA decay rather than production of truncated proteins (Gong et al., 2007). LQT4 is linked to loss-of-function mutations on the ANK2 gene encoding for ankyrin-B (Mohler et al., 2003,2004), a protein required for the proper localization of Na+/Ca2+ exchanger (NCX1), Na+/K+-ATPase (NKA), IP3 receptor (IP3R), and Cav1.3. The mutations cause an increase in intracellular Na+ facilitating afterdepolarizations and triggered arrhythmias under adrenergic stimulation (Ackerman and Mohler 2010). Mutations in the KCNE1 and KCNE2 genes encoding the auxiliary subunits MinK and MiRP1 are linked to LQT5 and LQT6, respectively (Abbott et al., 1999; Splawski, et al., 1997). Caveolin-3 is the main isoform expressed in the heart and coimmunoprecipitates with Nav1.5 subunits (Yarbrough et al., 2002). The LQTS9 is associated to mutations in the CAV3 gene encoding for caveolin 3 (Vatta et al., 2006), which results in a 2- to 3-fold increase in late sodium current with no changes in peak INa density and gating (Vatta et al., 2006). The LQT10 is linked to mutations in the SCN4B gene that encodes the β4-subunit of the Na+ channel (Medeiros-Domingo et al., 2007) and LQT11 to mutations on the AKAP9 gene (Chen et al., 2007) that encodes the Yotiao protein, a scaffolding protein that control the protein kinase A pathway. Yotiao modulates the response of IKs to α-adrenergic stimulation (Marx et al., 2002). The LQT12 variant is associated with mutations in the gene SNTA1 encoding for cardiac α1-syntrophin (Ueda et al., 2008; Chen et al., 2009), a protein linking the extracellular matrix to the intracellular cytoskeleton. Mutant SNTA1 releases inhibition of associated neuronal nitric oxide synthase by the plasma membrane Ca2+-ATPase (PMCA4b), leading to an increase in peak and late INa via S-nitrosylation of the cardiac Na+ channel. The LQT13 has been identified in a Chinese family carrier of a mutation in the KNCJ5 gene, encoding for the Kir3.4 subunit of the IKACh (Yang et al., 2010). The mutant protein interferes with the formation of functional KACh channels. A malignant form of LQTS associated to recurrent cardiac arrest due to VF in infants is associated to mutations in CALM1 and CALM2 genes encoding calmodulin (Crotti et al., 2013). Mutation carriers had recurrent cardiac arrest secondary to ventricular tachyarrhythmias, severely prolonged QTc interval (>600 ms), evidence of electrically unstable myocardium (T-wave alternans) and intermittent 2:1 AV block. Ventricular fibrillation was typically triggered by adrenergic activation, and either occurred spontaneously or was preceded by a short period of polymorphic VT that was not pause-dependent. Most carriers exhibited neurological deficits (epilepsy, neurodevelopmental delays) of variable severity that could be attributed to brain injury secondary to cardiac arrest or possibly to enhanced susceptibility to neuronal injury in the setting of circulatory insufficiency. Calmodulin serves as the Ca2+ sensor for Ca2+-dependent inactivation of L-type voltage-gated Ca2+ channels in cardiac myocytes (Peterson et al., 1999). Defects in calmodulin function may prolong repolarization because of impaired Ca2+-dependent inactivation of L-type calcium channels or dysregulation of voltage-gated Na+ channels leading to an increased depolarizing current during the plateau phase of the cardiac action potential.. Furthermore, the voltage-gated potassium channel KCNQ1 or KV7.1 responsible for the slow component of the delayed rectifier current (IKs) requires calmodulin for activity (Shamgar et al., 2006). Thus, inhibition of calmodulin inhibits IKs and delays repolarization setting the conditions for the appearance of early afterdepolarizations and triggered arrhythmias. Inactivation of cardiac Na+ channels also involves calmodulin and disrupting this interaction might evoke sodium channel dysfunction and conduction disturbances (Potet et al., 2009). Common variants can influence the QT interval. The NOS1AP gene that encodes nitric oxide synthase 1 adaptor protein (NOS1AP) is involved in cardiac repolarization. Variants of NOS1AP modulate the risk in LQTS and two intronic variants were associated with SCD even after controlling for QT interval (Deo et al., 2012). This association was validated to carry a 30 % increase in SCD risk in white participants, but not in African-Americans (Deo et al., 2012; George, 2009). The S1103Y variant in the SCN5A gene is common in African Americans (13 % being heterozygous) and increases the arrhythmia risk by more than 8-fold in carrier subjects (George, 2009). This variant produces an abnormal Na+ channel inactivation leading to increased susceptibility to early afterdepolarizations. In SCN5A-S1103Y homozygotes there is a 24-fold greater risk for ventricular arrhythmias and sudden death (George, 2009). 2. Jervell and Lange Nielsen syndrome. It is caused by homozygous or compound heterozygous mutations on either KCNQ1 or KCNE1 genes encoding the channel conducting IKs (Schwartz et al., 2006). Most mutations (80%) are on the KCNQ1 gene; mutations on the KCNE1 gene are associated with a more benign course. β-Blockers have only partial efficacy, so that 51% of the patients had events despite therapy and 27% had for cardiac arrest and SCD. Subgroups at relatively lower risk include females, patients with a QTc =550 ms, those without events in the first year of life, and those with mutations on KCNE1 (Schwartz et al., 2006). The LQTS has a low and highly variable penetrance (55% in the LQTS1, 70% and 80% LQTS2 LQTS3), which indicates that 2 out of 5 mutation carriers have a normal QTc (Priori et al.,1999, 2003). This happens even in patients with a history of syncope, although they are more susceptible to develop high-risk ventricular arrhythmias than the rest of the population. Some patients may remain asymptomatic for a long time, so that SCD can be the first clinical manifestation in a young "healthy" individual. Other patients present dizziness, lightheadedness (presyncope), syncope and seizures and MSC. Syncope can start early in life, often in infancy (before age 10, indicating a malignant course), but the frequency and intensity of symptoms may decrease to reach adulthood. When seizures occur in children they can be misdiagnosed of epilepsy. The occurrence of syncope during the performance of physical activity or strong emotion is a sign suggesting that the patient can present a LQTS. When there is also a family history of unexplained syncope or SCD the possibility that we are facing a SQTLc increases significantly. Thus, it is recommended to perform an ECG in all children and young people who have had an episode of syncope and/or unexplained seizures. Unfortunately, many times an electroencephalogram (EEG) is performed but not the ECG. Genotype/phenotype correlations in LQTS indicated that in LQT1 patients experience most of their symptoms during exercise (swimming, running o emotion); they should not practice in competitive sports. At rest, IKs has minimal activity, but this current becomes critical for shortening of the action potential at high heart rates and during adrenergic stimulation, which allows for shortening of the action potential duration with exercise (Schwartz et al., 2001; Terrenoire et al., 2005). An impairment of IKs can prevent appropriate shortening of the action potential duration during exercise, which may explain why exercise is a common trigger for sudden death in LQTS due to defects in the gene encoding IKs. Transmembrane mutations are related to longer QTc, more frequent cardiac events, and greater QTc prolongation with exercise (Jons et al., 2009). Cytoplasmic loop mutations that affect sites of adrenergically mediated phosphorylation are specifically associated with QT prolongation during exercise, but the QTc may be normal at rest. Mutations in the cytoplasmic loops of the KCNQ1 protein or mutations with dominant-negative ion current effects are associated with a worse prognosis, especially when compared with mutations affecting the C-terminal regions of the protein (Goldenberg et al., 2006; Jons et al., 2009). Loss-of-function mutations in KCNH2 result in a reduced IKr, delayed cardiac repolarization and QT prolongation. Patients with LQT2 experience events in association with increased sympathetic tone or emotional stress (unxpected noise: alarm clock, telephone, doorbell), postpartum or in patients with hypokalemia. In these patients are advised to remove the phones and alarm clocks in their bedroom (Schwartz et al., 2001). Most events associated with LQT3 (SCN5A mutations) occurred while the patients were asleep or at rest (55% of events) (Schwartz et al., 2001). Analysis of sex in relation to genetic subtype has revealed that females with LQT2 and males with LQT3 are at particularly high risk for sudden death or first cardiac arrest (Priori et al., 2003). When the mutant channel is expressed in other tissues, the LQTS is associated with extracardiac symptoms. Patients with Andersen - Tawil syndrome (SQTL7) caused by mutations in the gene KCNJ8 present QT prolongation, marked U waves, hypokalemic periodic paralysis, abnormal skeletal development, facial dysmorphic features (micrognathia, clinodactyly, hypertelorism, low set of ears) and and ventricular arrhythmias including bidirectional VT (Plaster et al., 2001; Yoon et al., 2011). Patients with Timothy syndrome (SQTL8) caused by gain-of-function mutations on CACNA1c gene encoding the a -subunit of the Cav1.2 channel, present marked QTc prolongation associated with facial malformations (round face, flat nasal bridge, receding upper jaw, syndactyly), congenital heart defects, atrioventricular block, intermittent hypoglycemia, cognitive disorders, autism and interdigital mergers and reduced immune response (Splawski et al., 2004). The missense mutation G406R, located in the cytoplasmic loop between domains I and II, produces maintained inward Ca2+ currents by causing nearly complete loss of voltage-dependent channel inactivation. This likely induces intracellular Ca2+ overload in multiple cell types. There is a relationship between age, gender, QTc duration, family history, genotype and clinical course, that allows risk stratification. Schwartz et al., (1993) published the diagnostic criteria for LQTS (Table), including electrocardiographic criteria, medical history and family history those subjects with = 3.5 points, have a high probability of LQTS.
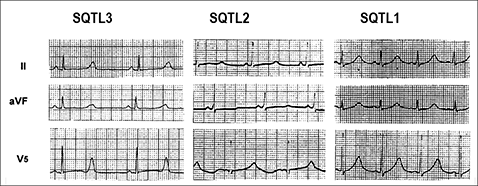
In patients with long QT syndrome, the ECG T-wave repolarization pattern is influenced by the genotype. Thus, in LQT1, the T wave is characteristically broad-based, while the T wave in LQT2 is typically bifid and of low amplitude. In contrast, in LQT3, prolongation of the ST segment accounts for QT prolongation and is typically followed by a a peaked, narrow T wave (Moss et al., 1995).
Hille B. Ion channels of excitable membranes. Hille B (Ed.). 2001. Payandeh J, Scheuer T, Zheng N, et al. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475:353-358. Cardiac Electrophysiology. From cell to beside. Eds. Zipes DP, Jalife J. Elsevier. Estados Unidos. Sixth Edition. 2013. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
| Aviso legal Esta obra está bajo una licencia de Creative Commons Reconocimiento-NoComercial-SinObraDerivada 4.0 Internacional |
