|
Diagnosis CPVT is a malignant inherited channelopathy caused an abnormal Ca2+ handling with onset during the pediatric age (Leenhardt et al., 1995). It is characterized by a normal ECG pattern at rest (although some patients present sinus bradycardia and a limit QT interval) and the appearance of bidirectional TV, syncope and SCD triggered by adrenergic stimulation, especially physical exercise or emotional stress in young patients with structurally normal hearts.
The diagnosis is mostly based on family history, symptoms and the detection of arrhythmias during exercise stress test or i.v. administration of catecholamines. During exercise or the i.v. administration of catecholamines (in patients who cannot perform an exercise test) CPVT patients develop ventricular extrasístoles at frequencies above 100-120 bpm, followed by an increase in the complexity ventricular extrasystoles and short runs of non-sustained VT. With continued exercise the duration of VT increases and a bidirectional VT (characterized by beat-to-beat 180º rotation of the QRS complex) appears that confirms the diagnosis (Napolitano et al., 2007). Some patients, however, can develop a polymorphic VT during the exercise test (Cerrone et al., 2009). Finally, FV, syncope and SCD appear. Once the exercise or stress test ends, the ECG changes reversed in the same way that occurred. Some CPVT patients also present supraventricular arrhythmias, mainly bursts of supraventricular tachycardia or atrial fibrillation that overlap with ventricular extrasystoles and VT. In asymptomatic relatives of CPVT patients the exercise test has a specificity of 97% and a sensitivity of 50% for predicting the presence of the familial CPVT-associated mutation. Role of ryanodine and calsequestrin CPVT is the result of an abnormality in the regulation of intracellular Ca2+ involving two main proteins located on the sarcoplasmic reticulum (SR): the ryanodine channel (RyR2) and calsequestrin. The cardiac RyR2 controls intracellular Ca2+ release and plays an important role in the excitation-contraction coupling. This process is initiated by the influx of extracellular Ca2+ following the activation of L-type Ca2+ channels during the lateau phase of the AP. This small entry of Ca2+ is insuffient to directly trigger a contractile response, but it can activate the RyR2 and facilitates the release of Ca2+ stored in the SR to the cytoplasm, leading to the activation of contractile proteins. This mechanism is known as Ca2+-induced Ca2+ release). During diastole, the [Ca2+]i decreases as a consequence of the activation of the Ca2+-ATPase pump (SERCA) that increases the uptake of Ca2+ into the SR, the Na+/Ca2+ exchanger located in the cell membrane (1 Ca2+:3 Na+) and the Ca2+-ATPase (PMCA) located in the cell membrane.
RyR2 is a homotetramer composed of four subunits of 4959 amino acids with a long cytoplasmatic N-terminal which extends intracytoplasmatic in the space between the SR and the T tubule membranes. Each monomer has 6 transmembrane segments forming the pore region of the channel. This N-terminal acts as a scaffold for regulatory subunits, enzymes, modulators (Ca2+, ATP, calmodulin) and drugs (caffeine and ryanodine) that regulate channel activity. Calsequestrin is the major Ca2+ fixing protein of 399 amino acids located in the SR and the release of calsequestrin-bound Ca2+ (through RyR2 channels) triggers muscle contraction. It also regulates the [Ca2+]i during the cardiac cycle, reducing cytoplasmic Ca2+ overload. Calsequestrin is the most abundant Ca2+ binding protein in the cardiac SR. CASQ2 normally limits RyR2 open probability and contributes to RyR deactivation after each Ca2+ release, so that it procides a large pool of Ca2+ releasable from the SR and at the same time limits RyR2 open probability and contributes to RyR deactivation after each Ca2+ release, so that it maintains the concentrations of free cytosolic Ca2+ at sufficiently low levels during the diastole.
CPVT is associated with mutations in RyR2, CASQ2, KCNJ2, TRDN, CALM1, and ANK2 genes which are implicated in cardiac intracellular calcium hemostasis. Mutations in RyR2 gene encoding the cardiac ryanodine receptor/Ca2+ release channel are found in approximately 65% of individuals and are inherited as an autosomal dominant disorder, with 80% penetrance (Laitinen et al., 2001; Piori et al., 2001; Tiso et al., 2001; Ackerman et al., 2011m, Rodriguez-Calvo et al., 2008; Modi et al., 2011). Mutations are located in certain regions of “hotspot areas” known as N-terminal (residues 77-466), central (2113-2534) and C-terminal domain (3778-4959) (Ackerman et al., 2011; Leenhardt et al., 2012). A deletion of exon 3 of RYR2 has been shown to cause a distinct subtype of CPVT characterized by sinoatrial node and atrioventricular node dysfunction, supraventricular arrhythmias, and dilated cardiomyopathy. A minority of cases (2-5%) result from recessive mutations in the cardiac calsequestrin isoform 2 (CASQ2) gene (Lahat et al., 2001; Di Barletta et al., 2006; Knollmann et al., 2006). Bilayer experiments demonstrated that removal of CASQ2 increases RyR2 open probability at fixed intraluminal Ca2+ suggesting that CASQ2 influences the open probability of RyR2 (Gyorke et al., 2004). CPVT2 is considered a more severe phenotype.
Mutations in the ANK-2 gene encoding ankyrin-2 and in the KCNJ2 gene encoding the potassium inwardly rectifying channel Kir2.1 have been reported in patients with exercise induced bi-directional VT (Mohler et al., 2004). Ankyrin-B mutations resulted in a loss of expression and abnormal coordination of NCX, Na+/K+-ATPase and the insositol-3-phosphate (InsP3) receptor. It has been also described an overlapping between TVPC and SQTL7, but its gravity is unknown. A novel missense mutation(I141V) in a highly conserved region of the SCN5A gene has been implicated in exercise-induced polymorphic ventricular tachyarrhythmias. The mutation shifted the activation curve toward more negative potentials and increased the window current (Swan et al., 2014). A yet-to-be-identified gene on chromosome 7p14–p22 (homozygous) has been linked to a highly malignant autosomal recessive form of CPVT (Bhuiyan et al., 2007). This phenotype is characterised by exercise-induced ventricular arrhythmia, and patients have a minor QT prolongation. The 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death (Priori et al., 2013) recommend:
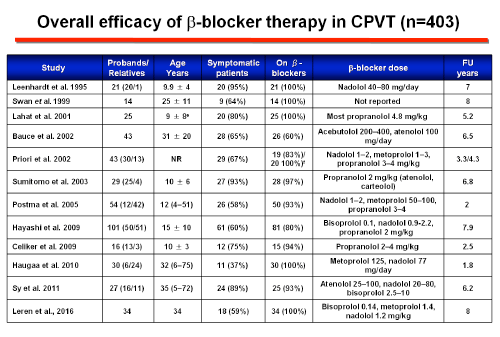
A robust comparison between different types of βBs has not been performed, but there is evidence that nadolol, a long-acting non-selective drug with a half-life of 12-24 hours, is the most effective βB in terms of ventricular arrhythmia suppression and cardiac event rates during exercise testing. Hayashi et al observed lower cardiac event rates in patients treated with nadolol compared with other β-blockers. However, this study did not indicate which type and dose of β-blocker the patients who were not on nadolol were receiving. Nadolol has a more pronounced chronotropic effect than other β-blockers and because of its long half-life (14-24 hours) offers the most stable, lasting degree of β-blockade and can be administered once daily (>1.5 mg/kg/day), which may result in improved adherence. Peopranolol, another nonselective β-blocker might be an alternative where nadolol is not available. The benefit of β-blockers has been attributed to their ability to block the adrenergic tone, but they can also modulate rate-dependent Ca2+ overload and reduce L-type Ca2+ channel current uring adrenergic stimulation. However, up to 30% of patients develop an arrhythmic event while on therapy (Napolitano and Priori, 2007). Patients who do not respond to treatment should be considered at very high risk.
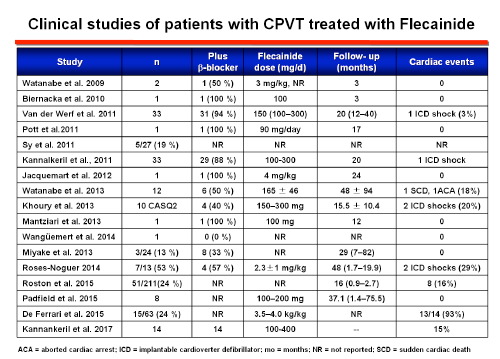
Current expert consensus guidelines have given a class IIa recommendation for the use of flecainide. The proposed underlying mechanism is: a) a direct blocking effect on voltage-gated sodium channels that raises the threshold for delayed afterdepolarization-induced triggered activity (Bannister ML et al. 2015; Liu N et al., Circ Res 2011; Sikkel MB et al., 2013), and b) the blockade of the open state of RyR2 channels (or an indirect effect on RyR2 through binding to calmodulin or other modulators of RyR2) that decreases the open probability of the channels, prevents RyR2-mediated premature Ca2+ release, inhibits early afterdepolarizations genesis and the onset of TV (Watanabe et al., 2009). RyR2-mediated sarcoplasmic reticulum Ca2+ regulates the beating rate of sinoatrial nodal cells in response to catecholamines. Flecainide reduces the rate of spontaneous SR Ca2+ release which may explain why maximum hearts rates are significantly lower in flecainide-treated patients even though workloads were higher compared with baseline exercise testing.
One small study evaluated the efficacy of flecainide as a monotherapy in 9 patients carrying RYR2 mutations either intolerant to or had experienced severe side effects of β-blockers (Padfield GJ et al., 2016). None of these patients suffered from treatment failure during a median follow-up of 37 months. However, even when flecainide monotherapy may potentially be considered an option for patients that are intolerant to β-blockers the routine use of flecainide monotherapy is not recommended because the evidence is scarce.
Ackerman MJ, Khositseth A, Tester DJ, et al. Epinephrine–induced QT interval prolongation: a genespecific paradoxical response in congenital long QT syndrome. Clin Proc 2002;77:413–21. Bannister ML, Thomas NL, Sikkel MB, et al. The mechanism of flecainide action in CPVT does not involve a direct effect on RyR2. Circ Res 2015;116:1324-1335. Bhuiyan ZA, Hamdan MA, Shamsi ET, et al. A novel early onset lethal form of catecholaminergic polymorphic ventricular tachycardia maps to chromosome 7p14-p22. J Cardiovasc Electrophysiol 2007;18:1060–1066 Biernacka EK, Hoffman P. Efficacy of flecainide in a patient with catecholaminergic polymorphic ventricular tachycardia. Europace. 2011;13:129-130. Cerrone M, Napolitano C, Priori SG. Catecholaminergic polymorphic ventricular tachycardia: a paradigm to understand mechanisms of arrhythmias associated to impaired Ca2+ regulation. Heart Rhythm 2009;6:1652–1659. Devalla HD, Gélinas R, Aburawi EH, et al. TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol Med. 2016;8:1390-1408. De Ferrari GM, Dusi V, Spazzolini C, et al. Clinical management of catecholaminergic polymorphic ventricular tachycardia: The role of left cardiac sympathetic denervation. Circulation 2015;131:2185-2193. Fernández-Velasco M, Rueda A, Rizzi N, et al. Increased Ca2+ sensitivity of the ryanodine receptor mutant RyR2R4496C underlies catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2009;104:201–9. Ferrero-Miliani L, Holst AG, Pehrson S, et al. Strategy for clinical evaluation and screening of sudden cardiac death relatives. Fundam Clin Pharmacol. 2010;24:619–35. Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J 2004;86:2121–2128 Hayashi M, Denjoy I, Extramiana F, et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation 2009;119: 2426–2434. Hwang HS, Hasdemir C, Laver D, et al. Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol 2011;4:128-35. Jacquemart C, Ould Abderrahmane F, Massin MM. Effects of flecainide therapy on inappropriate shocks and arrhythmias in catecholaminergic polymorphic ventricular tachycardia. J Electrocardiol 2012;45:736-738 Khoury A, Marai I, Suleiman M, et al. Flecainide therapy suppresses exercise-induced ventricular arrhythmias in patients with CASQ2-associated catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2013;10:1671-1675. Laitinen PJ, Brown KM, Piippo K, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001;103:485-90. Leenhardt A, Lucet V, Denjoy I, et al. Catecholaminergic polymorphic ventricular tachycardia in children. A 7–year follow–up of 21 patients. Circulation 1995;91:1512–9. Leenhardt A, Denjoy I, Guicheney P. Catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol 2012;5:1044-1052. Leren IS, Saberniak J, Majid E, et al. Nadolol decreases the incidence and severity of ventricular arrhythmias during exercise stress testing compared with β1-selective β-blockers in patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2016;13:433-440. Liu N, Denegri M, Ruan Y, et al. Flecainide exerts an antiarrhythmic effect in a mouse model of catecholaminergic polymorphic ventricular tachycardia by increasing the threshold for triggered activity [Short communication]. CircRes 2011;109:291-295. Liu N, Rizzi N, Boveri L, Priori SG. Ryanodine receptor and calsequestrin in arrhythmogenesis: what we have learn from genetic diseases and transgenic mice?. J Mol Cell Cardiol 2009;46:149–59. Mantziari L, Vassilikos V, Anastasakis A, et al. A de novo novel cardiac ryanodine mutation (Ser4155Tyr) associated with catecholaminergic polymorphic ventricular tachycardia. Ann Noninvasive Electrocardiol 2013;18:571-576. Maron BJ, Chaitman BR, Ackerman MJ, et al; Working Groups of the American Heart Association Committee on Exercise, Cardiac Rehabilitation, and Prevention; Councils on Clinical Cardiology and Cardiovascular Disease in the Young. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004;109:2807-2816. Miyake CY, Webster G, Czosek RJ, et al. Efficacy of implantable cardioverter defibrillators in young patients with catecholaminergic polymorphic ventricular tachycardia: Success depends on substrate. Circ Arrhythm Electrophysiol 2013;6:579-587. Mohamed U, Gollob MH, Gow RM, et al. Sudden cardiac death despite an implantable cardioverter-defibrillator in a young female with catecholaminergic ventricular tachycardia. Heart Rhythm 2006;3:1486–9. Modi S, Krahn AD. Sudden cardiac arrest without overt heart disease. Circulation. 2011;123:2994–3008. Mohler PJ, Splawski I, Napolitano C, et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci U S A. 2004;101:9137-42. Nyegaard M, Overgaard MT, Søndergaard MT, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91:703–712. Napolitano C, Priori SG. Diagnosis and treatment of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4:675–678. Olde Nordkamp LR, Driessen AH, Odero A, et al. Left cardiac sympathetic denervation in the Netherlands for the treatment of inherited arrhythmia syndromes. Neth Heart J 2014;22:160– Padfield GJ, AlAhmari L, Lieve KV, et al. Flecainide monotherapy is an option for selected patients with catecholaminergic polymorphic ventricular tachycardia intolerant of β-blockade. Heart Rhythm 2016;133:557 – 565. Postma AV, Denjoy I, Kamblock J, et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J Med Genet. 2005;42:863–70. Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001;103:196–200. Priori SG, Wilde AA, Horie M, et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 2013;15:1389–1406. Priori S.G., Blomstrom-Lundqvist C., Mazzanti A, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36:2757–2759. Roses-Noguer F, Jarman JWE, Clague JR, Till J. Outcomes of defibrillator therapy in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2014;11:58-66. Rosso R, Kalman JM, Rogowski O, et al. Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2007;4:1149–54. Roston TM, Vinocur JM, Maginot KR, et al. Catecholaminergic polymorphic ventricular tachycardia in children: Analysis of therapeutic strategies and outcomes from an international multicenter registry. Circ Arrhythm Electrophysiol 2015;8:633-642. Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. 2012;21:2759–2767. Sikkel MB, Collins TP, Rowlands C, et al. Flecainide reduces Ca(2+) spark and wave frequency via inhibition of the sarcolemmal sodium current. Cardiovasc Res 2013;98:286-296. Sumitomo N, Harada K, Nagashima M, et al. Catecholaminergic polymorphic ventricular tachycardia: electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart. 2003;89:66–70. Swan H, Laitinen P, Kontula K, et al. Calcium channel antagonism reduces exercise-induced ventricular arrhythmias in catecholaminergic polymorphic ventricular tachycardia patients with RyR2 mutations. J Cardiovasc Electrophysiol 2005;16:162–6. Swan H, Amarouch MY, Leinonen J, et al. A gain-of-function mutation of the SCN5A gene causes exercise-induced polymorphic ventricular arrhythmias. Circ Cardiovasc Genet. 2014;7:771–781. Tester DJ, Kopplin LJ, Will ML, et al. Spectrum and prevalence of cardiac ryanodine receptor (RyR2) mutations in a cohort of unrelated patients referred explicitly for long QT syndrome genetic testing. Heart Rhythm 2005;2:1099–105. Tiso N, Stephan DA, Nava A, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet 2001; 10:189-94. Uchinoumi H, Yano M, Suetomi T, et al Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ Res. 2010;106:1413–1424. van der Werf C, Kannankeril PJ, et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol 2011;57:2244–54. van der Werf C, Nederend I, Hofman N, et al. Familial evaluation in catecholaminergic polymorphic ventricular tachycardia: disease penetrance and expression in cardiac ryanodine receptor mutation-carrying relatives. Circ Arrhythm Electrophysiol 2012;5:748–756. Watanabe H, Chopra N, Laver D, et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med 2009;15:380–383. Watanabe H, Steele DS, Knollmann BC. Mechanism of antiarrhythmic effects of flecainide in catecholaminergic polymorphic ventricular tachycardia. Circ Res 2011;109:712–713. Watanabe H, van der Werf C, Roses-Noguer F, et al. Effects of flecainide on exercise-induced ventricular arrhythmias and recurrences in genotype-negative patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2013;10:542–7 Wilde AA, Bhuiyan ZA, Crotti L, et al. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. New Engl J Med 2008;358:2024–9. Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation 2006;114:e385–484.
|
|
|
| Aviso legal Esta obra está bajo una licencia de Creative Commons Reconocimiento-NoComercial-SinObraDerivada 4.0 Internacional |
