|
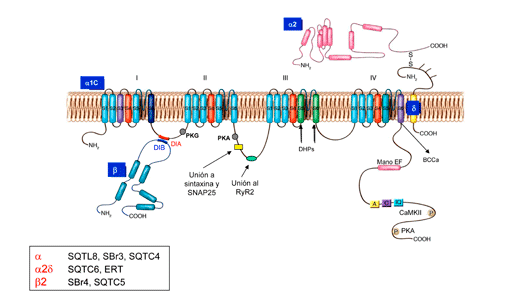
Calcium channel gating Channelopaties References In resting cardiomyocytes, the intracellular concentration of Ca2+ ([Ca2+]i) is 20,000 times less than its concentration in the extracellular environment (<0.1 m M vs 1-2 mM) and the cell interior is electronegative (-50 to-90 mV). Thus, there is an electrochemICal gradient that favors the entry of Ca2+ into the cell. When the cardiomyocytes is depolarized, the [Ca2+]i increases to 0.3-1 m M due to the entry of extracellular Ca2+ through L-type channels of the sarcolemma (and to a much lesser extent via the na+-Ca2+ exchanger) and/or the release of Ca2+ from intracellular stores, mainly the sarcoplasmic reticulum (Ca2+ induced Ca2+ release). The cardiac L-type calcium channel is a hetero-oligomeric protein consisting of a pore-forming α1 sununit (α1C, 200 kDa), which is able to form functional channels, and a set of auxiliary or regulatory subunits: a disulfide-linked subunit dimer a 2/ d (175 kDa) and intracelular β1-3 (55-60 kDa) subunits (Figure) which are encoged by the genes CACna1C (or CACna1D), CACNA2D1 (or CACNA2D3) and CACNB1-3, respectively (Nerbonne y Kass, 2005; Bodi y cols., 2005) (Figure). β Subunits are tightly associated at the cytoplasmic face of α1 (through the I–II linker), whereas α2δ subunits are GPI-anchored to the plasma membrane and interact with extracellular domains of α1. The α1C subunit (Cav1.2, 2179 amino acids) consists of four homologous domains (I-IV), each with six transmembrane segments (S1-S6), which associate in the cytoplasmatic membrane to form the ion-conducting pore. This subunit also contains the voltage sensor of the channel, comprised by transmembrane segments S1–S4 in each homologous repeat (I–IV), the pore-forming region comprised by S5 and S6 segments together with their connecting linker, which contains helical regions contributing to the formation of the selectivity filter. A Ca2+-selective pore region is located between S5 and S6 (Caterall, 2000). The high (100-fold) selectivity of the channel for Ca2+ over small monovalent cations is related to a conserved ring of glutamates (EEEE) located in the pore that affinity Ca2+ (KD =500 nmol/L) forming the filter channel selectivity (Koch, et al., 2000). The interaction between a 1 and the cytoplasmic b subunit is mediated by a well-defined sequence motifs on b (BID, b subunits interaction domain) and on the I-II cytoplasmic linker of a 1 (AID). The possible interaction sites between the a subunit and the ryanodine receptor (RyR2) of the sarcoplasmic reticulum are localized in the linker between DII and DIII. The α1C subunit also harbors the binding sites for channel-modulating drugs.
The β subunits (55 kDa) are cytosolic proteins that assemble with α1-subunits and regulate the expression of functional cell surface L-type Four different β-subunit-encoding genes, CACNB1, CACNB2, CACNB3, and CACNB4, which encode the β1-4 subunits, respectively, have been identified. In general, coexpression of β subunits modulates the biophysical properties of the α1 subunits, producing a leftward shift of the current-voltage relationship, which is consistent with the involvement of the S4 region of the α1 subunit voltage-sensor region. The α2δ subunit (143 kDa), encoded by CACNA2D1, CACNA2D2, CACNA2D3, and CACNA2D4 genes, is transcribed from a single gene, translated and proteolytICally processed into α2 (N-terminus) and δ (C-terminus, extracellular) subunits that remain linked through disulfide bonds. The α2 domain is located extracellularly, and the δ subunity has a single transmembrane region with a very short intracellular part. Coexpression of α2δ with α1C subunit causes a 2-fold increase in expression of dihydropyridine binding sites, gating currents and ICa(Nerbonne y Kass, 2005), shifts the voltage dependence of activation of α1-β encoded channels and accelerates current activation and inactivation (Bangalore et al., 1996; Gurnett et al., 1996; Singer et al., 1991). It is probable that the α2/δ and β subunits “drive” the α1C subunit to the membrane in the correct insertion mode.
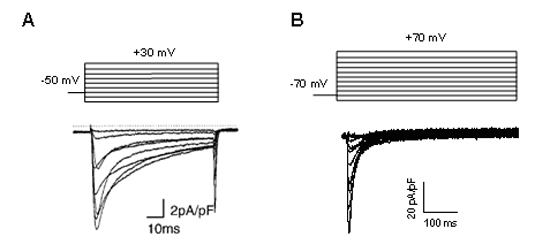
Figure. L-type Ca2+ current recorded in human atrial cardiomyocytes.
L-type Ca2+ channels activate-open on depolarization to membrane potentialspotentials positive to approximately -40 mV and the current amplitude peaks at around 0 mV. The channels activated very rapidly during depolarization (reaching a peak withing 2-7 ms) and remained open during the plateau phase of the AP generating the ICa. Channels inactivate over a time course of several tens of milliseconds to seconds. Theoverlap between the activation and inactivation curves indicates a “window current”. When the cardiac AP are very long (prolonged QT interval), as the membrane potential enter in this region (between -15 and -40 mV) the ICathat was largely inactivated during the plateau phase can be partly reactivated as the [Ca2+]i declines. This reactivation can cause a net depolarization inducing early depolarizations and triggered focal activity.
Figure.ICa, L recorded in human atrial cardiomyocytes. Mutations in the CACna1C gene encoding Cav1.2 are associated to LQT8 (Tymothy syndrome), BrS3, SQTS4 and ERS2, mutations in the CACNA2D1 encoding the a2d subunit in the SQTS6 and in the ERS4, mutations in the CACNB2B gene encoding the b2 subunit in the SBr4, the SQTS5 and the ERS3. Bangalore R, Mehrke G, Gingrich K, et al. Influence of L-type Ca channel alpha 2/delta-subunit on ionic and gating current in transiently transfected HEK 293 cells. Am J Physiol Heart Circ Physiol 1996; 270:H1521-H1528. Bezanilla F. Voltage sensor movements. J Gen Physiol 2002;120:465-473. Bichet D, Cornet V, Geib S et al. The I-II loop of the Ca2+ channel alpha1 subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron. 2000;25:177-190. Bodi I, Mikala G, Koch SE, et al. The L-type calcium channel in the heart: the beat goes on. J Clin Invest. 2005;115:3306-3317. Caterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011;3:a003947 Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000; 16:521-555. Davies A, Hendrich J, Van Minh AT, et al. Functional biology of the alpha2-delta sununits of voltage-gated calcium channels. Trends Pharmacol Sci 2007;28:220-228. Dolphin AC. G protein modulation of voltage-gated Calcium channels. Phamacol Rev 2003;55:607-627. Gurnett CA, De Waard M, Campbell KP. Dual function of the voltage-dependent Ca2 _ channel alpha 2 delta subunit in current stimulation and subunit interaction. Neuron 1996;16:431-440. Hering S, Berjukow S, Aczel S, et al. Ca2+ channel block and inactivation: common molecular determinants. Trends Pharmacol. Sci. 1998; 19:439-44 Hockerman GH, Peterson BZ, Johnson BD, et al. Molecular determinants of drug binding and action on L-type calcium channels. Annu Rev Pharmacol Toxicol. 1997; 37:361-396. Hulme JT, Lin TW, Westenbroek RE, et al. Beta-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc natl Acad Sci U S A. 2003;100:13093-8. Hulme JT, Yarov-Yarovoy V, Lin TW, et al. Autoinhibitory control of the CaV1.2 channel by its proteolytICally processed distal C-terminal domain. J Physiol. 2006;576:87-102. Kass RS, Sanguinetti MC. Inactivation of calcium channel current in the calf cardiac Purkinje fiber. Evidence for voltage-and calcium-mediated mechanisms. J Gen Physiol 1984;84:705-726 Kim J, Ghosh S, Nunziato DA, et al. IdentifICation of the components controlling inactivation of voltage-gated Ca2+ channels. Neuron 2004a;41:745-754. Kobrinsky E, Tiwari S, Maltsev VA, et al. Differential role of the alpha1C subunit tails in regulation of the Cav1.2 channel by membrane potential, beta subunits, and Ca2+ ions. J. Biol. Chem 2005;280:12474–12485. Koch SE, Bodi I, Schwartz A, Varadi G. Architecture of Ca2+ channel pore-lining segments revealed by covalent modifICation of substituted cysteines. J Biol Chem 2000;275:34493-34500 Lee KS, Marbán E, Tsien RW. Inactivation of calcium channels in mammalian heart cells: joint dependence on membrane potential and intracellular calcium. J Physiol (Lond.) 1985;364:395-411. Mikala G, Klockner U, Varadi M, et al. cAMP-dependent phosphorylation sites and macroscopic activity of recombinant cardiac L-type calcium channels. Mol Cell Biochem 1998;185:95-109. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev 2005;85:1205-125 Perez-Reyes E, Castellano A, Kim HS, et al. Cloning and expression of a cardiac/brain beta subunit of the L-type calcium channel. J Biol Chem 1992; 267:1792-1797. Pragnell M, De Waard M, Mori Y, et al. Calcium channel betasubunit binds to a conserved motif in the I-II cytoplasmic linker of the alpha 1-subunit. nature 1994;368:67-70. Qin N, Olcese R, Bransby M, et al. Ca2+-induced inhibition of the cardiac Ca2+channel depends on calmodulin. Proc natl Acad Sci 1999;96: 2435–2438. Singer D, Biel M, Lotan I, et al. The roles of the subunits in the function of the calcium channel. Science 1991;253:1553–1557. Viard P, Butcher AJ, Halet G, et al. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. nat. Neurosci. 2004;7:939–946. Wei SK, Colecraft HM, DeMaria CD, et alT. Ca2+ channel modulation by recombinant auxiliary beta subunits expressed in young adult heart cells. Circ Res 200; 86:175-184. Yamaguchi H, Hara M, Strobeck M,et al. Multiple modulation pathways of calcium channel activity by a beta subunit. Direct evidence of beta subunit participation in membrane trafficking of the alpha1C subunit. J Biol Chem 1998; 273:19348-19356. Zuhlke RD, Pitt GS, Deisseroth K, et al. Calmodulin supports both inactivation and facilitation of L-type calcium channels. nature 1999;399:159-162. |
|
|
| Aviso legal Esta obra está bajo una licencia de Creative Commons Reconocimiento-NoComercial-SinObraDerivada 4.0 Internacional |
