|
Classification
Cardiac K+ channels are the primary determinants of action potential repolarization, determine the shape and duration of the action potential, the resting membrane potential, the heart rate, and are important targets for the actions of neurotransmitters, hormones, drugs and toxins known to modulate cardiac function (Tamargo et al., 2004; Nerbonne and Kass 2005).
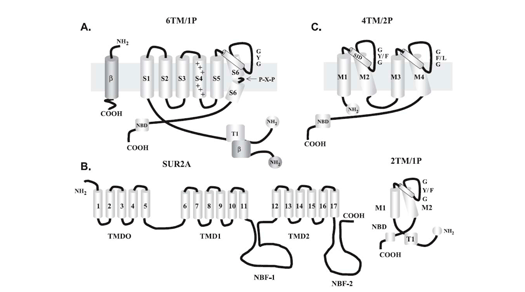
Cardiac K+ currents can be distinguished on the basis of their topology and differences in their functional and pharmacological properties. K+ channels are formed by coassembly of pore-forming a -subunits which surround a water-filled pore and accessory b -subunits and with additional associated proteins, such as regulatory enzymes or cytoskeleton proteins. Over 80 human K+ channel-related genes have been cloned and characterized (Alexander et al., 2011; Coetzee et al., 1999; Jenkinson et al., 2006; Lüscher y Slesinger 2010; Nerbonne y Kass 2005; Tamargo et al., 200 4). The genes encoding the α- and β-subunits of cardiac K+ channels are shown in Table 1.
In mammalian cardiac cells, K+ channels can also be categorized as voltage-gated (Kv) and ligand-gated channels
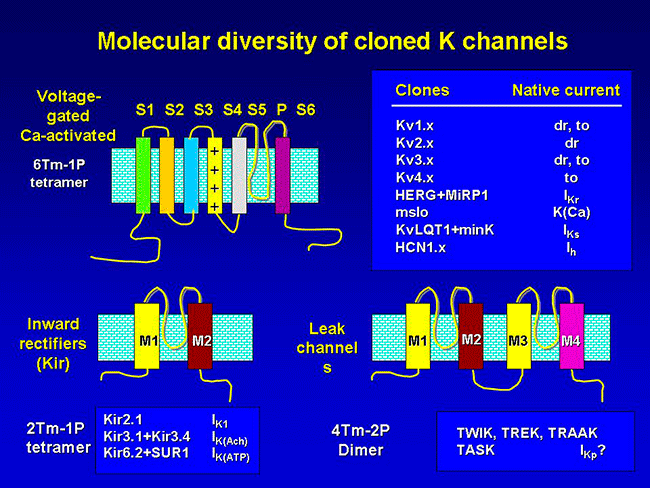
1. CHANNELS CONTAINING SIX TRANSMEMBRANE SEGMENTS AND ONE PORE-FORMING REGION (6TM-1P)
Kv channels are formed by the tetrameric coassembly of pore-forming α-subunits which surround a water-filled pore and accessory β -subunits. Channels can be formed from four identical a-subunits (homomultimers) or from combinations of different a -subunits from the same subfamily (heteromultimers).
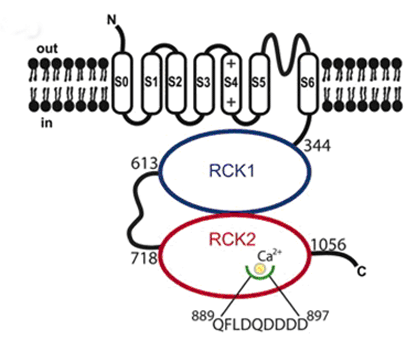
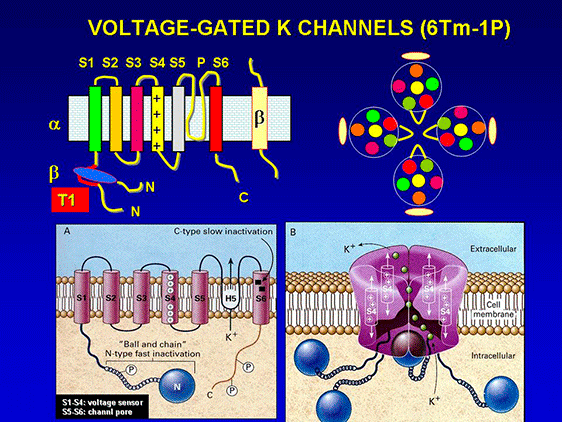
Kv channel α-subunits have six transmembrane-spanning segments (S1-S6) with cytoplasmic N- and C-terminal domains and a pore loop between S5 and S6 bearing the K+ selectivity filter signature TVGYG (Caterall, 2010). K+ ions are conducted very efficiently, at near diffusion-limited rates (107 ions channel−1s−1) and simultaneously, K+channels are highly selective and at least 10,000 times more permeant for K+ than Na+ (Doyle et al., 1998, Sansom et al., 2002). The pore region, formed by S5 and S6 segments and the S5-S6 linker (P loop), is responsible for K+ ion conduction and selectivity. The P region comprises 19 amino acids (the first 8 and the last 3 residues are located at the entrance to the pore) and penetrates the interior of the membrane; it also contains the largely conserved sequence [(T/S)xxTxGYG] which determines K+ selectivity (MacKinnon 1991; Heginbotham et al., 1994). The S4 segment contains positively charged residues (arginine or lysine) at approximately every third position and together with segments S1-S3 serves as the voltage sensor. The inner mouth of the channel is formed by the segment connecting S4 and S5 and the cytoplasmic regions of S5 and S6, while the outer mouth is formed by the P region and the extracellular part of the segments S5 and S6. The segment connecting S4- S5 also forms part of the receptor for the inactivation particle. The voltage sensor is formed by the segment S4, wich presents ne positively charged amino acid every three residues [XX -(A / L)] and the negative charges of the segments S2 and S3, which exert an electrostatic action with S4 (Liman et al., 1991; MacKinnon 1991; Pongs, 1992). Mutations that neutralize the charged amino acids in S4 move the midpoint value channel activation, which confirms its role in the conformational change leading to the opening (Liman et al., 1991; Papazian et al., 1991). The α subunit of high conductance Ca2+ -activated K+ channels (BKCa) The α subunit of high conductance Ca2+ -activated K+ channels (BKCa) is encoded by the KCNMA1 gene. The α subunit presents a short extracellular N-terminus, 7 transmembrane segments (S0-S6). S1-S4 forms the voltage sensor (S4 contains several Arginine residues which act to ‘sense’ the changes in charge and move in a very similar way to other Kv channels) and S5 -S6 the channel pore that contains the selectivity filter) and a long intracellular C-terminal domain (CTD) which contains two RCK (motifs regulating the conductance of K+), the second of which contains the binding sites for Ca2+ (Yuan et al. Science 2010) (Figure). β subunits encoded by genes KCNMB1-4 assembled with α subunits forming tetrameric channels. β1 and β2 subunits alter Ca2+ dependency; β3 alters channel gating (facilitating its inactivating) and β4 modulates the phosphorylation subunits. The α subunit of high conductance Ca2+ -activated K+ channels (BKCa) is encoded by the KCNMA1 gene. The α subunit presents a short extracellular N-terminus, 7 transmembrane segments (S0-S6). S1-S4 forms the voltage sensor (S4 contains several Arginine residues which act to ‘sense’ the changes in charge and move in a very similar way to other Kv channels) and S5 -S6 the channel pore that contains the selectivity filter) and a long intracellular C-terminal domain (CTD) which contains two RCK (motifs regulating the conductance of K+), the second of which contains the binding sites for Ca2+ (Yuan et al. Science 2010) (Figure). β subunits encoded by genes KCNMB1-4 assembled with α subunits forming tetrameric channels. β1 and β2 subunits alter Ca2+ dependency; β3 alters channel gating (facilitating its inactivating) and β4 modulates the phosphorylation subunits.
K+ channels usually present three conformational configurations: an open state (O), and two non-conductive, closed (C) and inactive (I) states (Zagotta and Aldrich, 1990a, b). Immediately after depolarization Kv channels move from the closed to the open state (activation) with a sigmoidal kinetics suggesting that must move through various non-conductive states, first to a closed state from which the openable channel (C*) and then from C* to O: C ↔ C ↔ Cn ↔ C* ↔ O ↔ I.
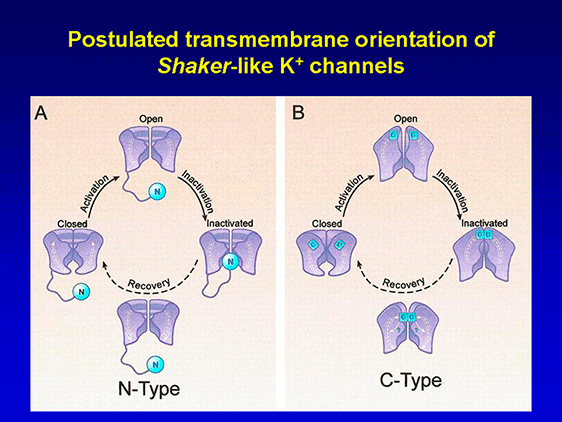
Channel activation involves relatively large displacements of S4 leading to channel opening by mean of direct coupling between the S4-S5 linker region and the C terminal end of S6, which with S5 lines the intracellular mouth of the channel pore (Yellen, 2002). The C-terminal segment of S6 contains a highly conserved sequence (PxP or PxG), which is critical in coupling the movement of the voltage sensor to channel opening. Then, many channels inactivate (turn to the nonconductive states), leading to a decline in activated macroscopic current (Jiang et al., 2003). The inactivation of the channel can be performed from the open state (O → I) or without channel openning (C* → I), while the deactivation (I ↔ O) takes place when the membrane is hyperpolarized. When the membrane is repolarized channels recover from the inactivated state back to the closed state (I → C) and are once again capable of opening in response to membrane depolarization. There are two major types of inactivation (Yellen, 2002; Imai et al., 2010). The N-type is a fast autoinhibitory process present in channels that generate transient currents (Kv1.1 and Kv1.4, Kv4.2 and Kv4.3, Kv3.1-3.4 and KCNM) results from the occlusion of the intracellular mouth of the pore by a ‘‘ball and chain’’ mechanism when the channel opens (Coetzee et al., 1999; Rassmusson et al., 1998). In this model, the first three amino acid residues at the N-terminus (inactivation ball) bind to the central cavity ("ball of inactivation") via an amino acid chain (residues 23-83) to a negatively charged receptor localized in the central cavity and inner pore of the K+ channel, the following eight hydrophobic amino acid residues extend from the cavity to the intracellular entryway, and the subsequent nine hydrophilic amino acid residues interact with the aqueous protein surfaces of the cytosolic domain (Zhou et al. 2001). The rapid (1-10 ms) inactivation is abolished by intracellular administration of TEA and mutations that suppress the first 22 amino acids of the N-terminal sequence or break the hydrophobic segments S4, S5 and it can be recovered after adding the removed peptide (Hoshi et al., 1991, Lopez-Barneo et al., 1993; MacKinnon and Yellen, 1990). The C-type slow inactivation of Kv1.2 channels, Kv1.5, Kv2.1 and KCNH2 occurs at the external outer mouth of the pore and appears to result from conformational changes of the SF, together with elimination of K+ ions and water molecules (Imai et al., 2010; Cuello et al., 2010). The residues alanine469 in the S6 segment and threonine (Thr449) located in the extracellular portion of P loop play a key role in this type of inactivation (Hoshi et al., 1990; Pong et al., 1992, Lopez-Barneo et al., 1993). C-type inactivation is suppressed by mutations in residues located in the extracellular mouth of the channel and in the extracellular portion of segment S6 and is delayed after applying TEAo and by increasing [K+ ] o, due to the interaction of K+ with a high affinity site located on the outer side of the ion channel pore (López- Barneo et al., 1993). Although the expression of the a subunits is sufficient to generate functional K+ channel, its coexpression with accessory subunits is necessary to reproduce the characteristics of native channels. The coexpression of a and β subunits r egulates cell surface expression, gating kinetics and drug-sensitivity of K+ channels (Hanlon and Wallace, 2002). K+ channel b -subunits represent a diverse molecular group, which includes:
Kv β1.1-1.3 subunits associate with the a subunits of Kv1 channels subunits and Kv β4 with the Kv2 channels. Each β subunit contains a conserved core region with a variable N-terminus. In Shaker channels the interaction between α and β subunits occurs between the domains located in the N-terminal of the α subunit and the C- terminal of the β subunit (Wang et al. 1996c; Gulbis et al., 2000).
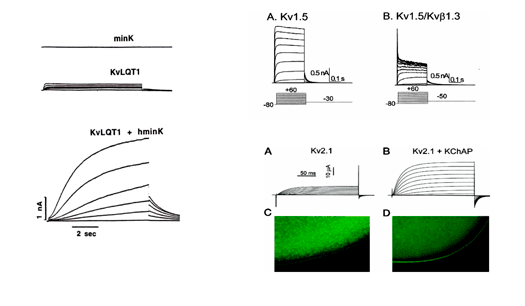
MinK coasesembly with different α (Kv7.1, Kv11.1 y Kv4.3) subunits; so, it can regulate IKs, IKr and Ito. MiRP (MinK-Related Peptides ) subunits can coassemble with Kv11.1 (Barhanin et al., 1996; Sanguinetti et al., 1996), Kv7.1 (McDonald et al., 1997) and Kv4.2 subunits and, thereby, regulate IKs, IKr and Ito (McCrossan y Abbott, 2004). MiRP1 (encoded by the KCNE2 gene) function as an accessory subunit of HERG1 (encoded by the KCNH2 gene) to generate cardiacIKr (Abbott y Golstein, 2001). In heterologous systems the MiRP subfamily of Kv channel accessory subunits can assemble with Kv α-subunits in several different subfamilies (Abbott et al., 2001; Zhang et al., 2001) to modify the properties and/or the cell surface expression of α-subunit-encoded channels. MiRP1 can also associate with hyperpolarization-activated cyclic nucleotide-gated (HCN) cation channels, which suggests a role for the MiRP1 subunit in the regulation of myocardial pacemaker currents (Yu et al., 2001).
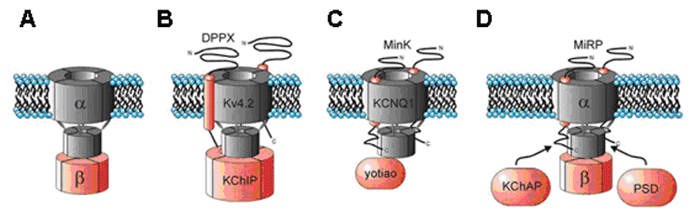
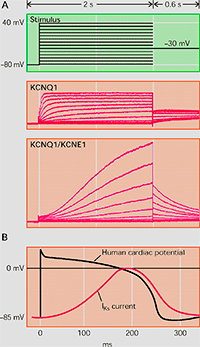
Figure. Ion channel subunits. (A) Assembly of the Kv channel α subunit through the N- terminal to a tetramer cytoplasmic β subunits. (B) Interaction of Kv4.2 channel α subunit to 4 cytoplasmic KCHIP subunits and one DPPX subunit. (C) Association of Kv7.1 channel with 2 MinK subunits to generate IKs. We can see the macromolecular channel complex with the yotiao adapter protein, which in turn binds to regulatory proteins of protein kinase A and protein phosphatase 1. (D) A complex formed by an α subunit of the Kv11.1 channel that generates the IKr, the regulatory subunits MIRP and KChAP and a postsy naptic density protein (PSD). Adaptada de McCrossan y Abbott, 2004. The rapidly activating componente of the delayed rectifier K+ current (IKr) The rapidly activating component (IKr) of the delayed rectifier K+ current contributes to the phase 3 of cardiac repolarization and plays an importante role in the duration of the cardiac AP and refractoriness. The native IKr results from the coassembly of the Kv11.1 a subunit (hERG encoded by KCNH2) with different two ancillary β -subunits, MinK (encoded by the KCNE1 gene) and MiRP1 (encoded by the KCNE2 gene) (Sanguinetti). In humans, KCNH, the gene encoding the human ether a go-go-related gene type 1 (hERG1) K+ channel, is located on chromosome 7q35–36, and the coding region comprises 16 exons spanning approximately 34 kb of genomic sequence. The full-length hERG1 subunit is composed of 1,159 amino acids with a predicted molecular mass of 127 kDa and has six transmembrane domains (S1–S6). hERG1a has a long (376 amino acids) N-terminus and residues 1-135 contains a protein-protein interaction structure called a Per–Arnt–Sim (PAS) domain which normally interacts with the channel and reduces the rate of deactivation (Morais et al., 1998). hERG1a has a long (376 amino acids) N-terminus and contains a protein-protein interaction structure called a Per-Arnt-Sim (PAS) domain which normally interacts with the channel and reduces the rate of deactivation (Morais et al., 1998). The PAS domain can be phosphorylated (Cui et al., 2000) and needs to be properly folded for the normal trafficking of the channel complex from the endoplasmic reticulum (ER) to the Golgi and cell surface (Paulussen et al. 2002). The slow deactivation associated with the hERG1a subunits may be mediated by an interaction between the PAS domain and the S4-S5 linker (Wang et al., 1998), a structure that couples the voltage sensor movement to opening and closing of the activation gate (Long et al., 2005). LQTS associated mutations in this region disrupt channel trafficking and its deletion greatly accelerates the rate of deactivation (Chen et al., 1999; Morais et al., 1998), perhaps by disrupting its interaction with the S4-S5 linker of the channel (Sanguinetti, 2010). The C-terminal contains a cyclic nucleotide-binding domain (CNBD). HERG channels are regulated by PKA at phosphorylation sites localed in the C- and N-termini, and by cAMP at the CNBD. PKA activation reduces HERG currents, while direct binding of cAMP to the HERG channel increases the current amplitude. cAMP increases the current when HERG is coexpressed with MiRP1 or MinK, consistent with the finding that loss-of-function mutations in the CNBD are associated with the LQT2 syndrome (Satler et al., 1996; Tseng, 2001). Homozygous mutations in KCNH2 are very rare as they result in intrauterine death or live birth with severe QT prolongation (Hoorntje et al., 1999; Johnson et al., 2003). Mutations in KCNH2 causing LQT2 can result in a reduction in IKr by dominant-negative suppression (pore mutations), nonfunctional channels or loss of function related to deffects in channel trafficking to the plasma membrane (Sanguinetti 2010; Zhou et al., 1998; Splawski y cols., 2000; Kass y Moss, 2003; Sanguinetti y Tristani-Firouzi, 2006; Sanguinetti 2010). Slow component of the delayed rectifier (IKs) KCNQ1 gene encodes the a-subunit of the slow componentof delayed rectifier K+ current (IKs) channel (Kv7.1).This protein, together with the b-subunit KCNE1 and an adapter protein Yotiao, forms a macromolecular complex (i.e., the functional potassium ion channel IKs) (Barhanin y cols., 1996; Marx et al., 2002; Sanguinetti y cols., 1996). The LQT1-related KCNQ1 gene is 404 kb long and located on chromosome 11p15.5. This gene codes for a 75-kDa protein containing 676 aminoacids (Wang et al., 1996). Each subunit contains six membrane-spanning segments (S1–S6 involving aminoacid residues 122– 348) connected by alternating intra- and extra-cellular loops, as well as a pore loop (aminoacid residues 300–320) located between segments S5 and S6, with a cytosolic N-terminus (1–121) and a long cytosolic C-terminus (residues 349-676) (Wu et al., 2016). The four subunits form a symmetrical alignment for the channel molecule together with KCNE1 (129 amino acids with a single transmembrane segment) and Yotiao proteins, and construct a specialized pathway that allows for the conduction of potassium ions through water-filled pores located in the center of the complex. S1-S4 segments of the potassium channel form a voltage-sensing domain (VSD). The pore region is composed of two transmembrane segments (S5-S6) joined together by a linker (including a pore loop) that contains the conserved aminoacids of the selectivity filter(residues 312–317) and affects the channel current amplitude, ion selectivity and channel blockade (Kurokawa et al., 2001; Wu et al., 2016). KCNQ1 possesses a large COOH terminus that is important for channel gating, assembly, and trafficking (Dvir et al., 2014; Wu et al., 2016). The C-terminus is comprised of four amphipathic a-helices, coiled-coils, and clusters of basic aminoacids. In the C-terminus, a leucine zipper motif (residues 588–616) is a unique site through which A-kinase anchoring protein 9 (AKAP9, or Yotiao) targets protein kinase A (PKA) and protein phosphatase 1 (PP1) to the KCNQ1 complex (Marx et al., 2002).
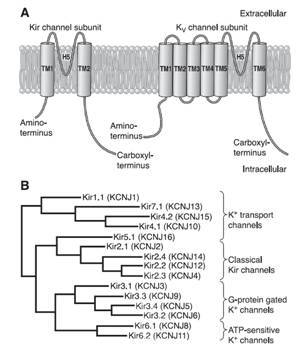
β-adrenoceptor stimulation increases protein kinase A (PKA) phosphorylation of IKs leading to a marked increase in current amplitude, by increasing the rate of channel activation and reducing the rate of channel deactivation (Marx et al., 2002; Furokawa et al., 2003). A leucine zipper motif in the C-terminus of KCNQ1 coordinates the binding of a targeting protein yotiao (Clancy and Kass, 2005), which in turn binds to and recruits PKA and protein phosphatase 1 (PP1) to the channel. The complex then regulates the phosphorylation of Ser- 27 in the N-terminus of KCNQ1 (Marx et al., 2002). PKA phosphorylation of IKs considerably increases current amplitude by increasing the rate of channel activation (C → O) and reducing the rate of channel deactivation (O → C) leading to increased current ampliutde and faster cardiac repolarization (Furokawa et al., 2003; Marx et al., 2002). Mutations in KCNQ1 can cause dysfunction in the IKs channel, such as a delay in channel opening or a reduction in the duration for which it is open (Keating et al., 1991; Moss et al., 2007; Dvir et al., 2014). This results in a decrease in repolarizing K+ current or a loss-of-function during phase 3 of the cardiac AP, which eventually causes QT prolongation and serious arrhythmias. LQT1 is more frequently triggered by adrenergic stimuli (e.g., physical exertion or emotional stress) compared with other forms of LQTS, particularly by diving and swimming (Schwartz et al., 2001; Ackerman et al., 1999) Mutations in KCNQ1 or KCNE1 can reduce IKs through dominant negative effects, reduced responsiveness to β-AR signaling (Abbott and Goldstein, 2002; Kurokawa et al., 2003), or alterations in channel gating (i.e, a reduction in the rate of channel activation or an increase in the rate of channel deactivation) (Bianchi et al., 1999; Chen et al., 1999; Chouabe et al. 2000; Splawski et al., 2001). Mutations in KCNQ1 cause LQTS by a reduction in or a complete loss of current, resulting in reduced repolarizing current during the AP (Wang et al., 1996). These mutations often act by dominant negative suppression of normal subunits, which results in a significant reduction in current. In other instances, there is a complete loss of current, which has been demonstrated to result from the assembly of nonfunctional channels or the failure of channels to traffic to the plasma membrane (Bianchi et al., 1999). Mutations can also alter channel-gating properties, which typically manifest as either reduction in the rate of channel activation (R539W, R555C) (Chouabe et al., 1997, 2000), or an increased rate of channel deactivation (including (S74L, V47F, W87R, KCNE1 and W248R KCNQ1) (Franqueza et al., 1999; Bianchi et al., 1999; Splawski et al., 2001). A LQTS-associated KCNQ1 COOH-terminal mutation, G589D, disrupts the leucine zipper motif and prevents cAMP-dependent regulation of IKs (Marx et al., 2002). The reduction of sensitivity to sympathetic activity likely prevents appropriate shortening of the APD in response to increases in heart rate. 2. CHANNELS WITH TWO TRANSMEMBRANE DOMAINS AND ONE PORE (2 TM-1P) Inwardly rectifying K+ channels encoded by KCNJ1-15 genes carry inward K+ currents better than outward K+ currents over a limited range of membrane potentials (Lopatin and Nichols, 2001). They conduct K+ currents more in the inward direction than the outward and play an important role in setting the resting potential close to the equilibrium potential for K+ (approximately ~ - 90 mV). There are seven Kir channel subfamilies (Kir1.x-Kir7.x, where x is the number of each member) that can be classified into four functional groups based on their mediators and the properties of ion conduction (McKinnon 2003; Hibino et al., 2010):
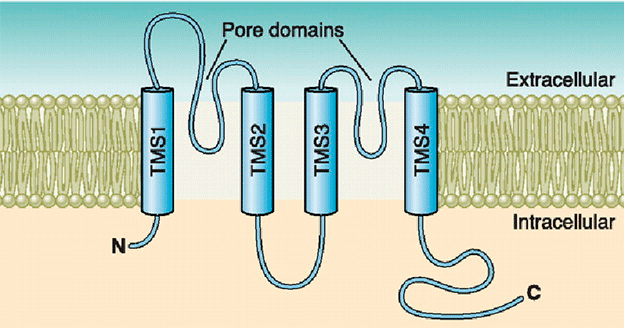
Inward rectifier K+ channels (Kir1.x-Kir7.x) contain two transmembrane domains (M1 and M2, similar to S5 and S6 in the Shaker family) connected by a pore-forming loop containing the G(Y/F)G sequence and cytosolic N- and C-termini (Coetzee et al., 1999; Nerbonne and Kass, 2005; Tamargo et al., 2004; Lopatin and Nichols, 2001; Seino and Miki, 2003; Hibino et al., 2010). The pore-forming domain is responsible for ion conduction while the cytosolic domain regulates the gating of the channel. Kir channels form either homo- or heterotetramers. The C-terminal participates in the ionic selectivity and internal rectification of the channel and possess a class I PDZ domain recognition sequence (X-S/T-X-V/I) that allows its binding to several anchoring proteins (PSD-95/SAP90). The cytosolic domain forms a binding site to interact with diverse intracellular regulatory mediators (Hibino et al., 2010). Multiple ion binding sites (D173, E225, D256, and E300 in Kir2.2) in this domain are conserved and critical to inward rectification. In addition to the intracellular and extracellular gates in the transmembrane part, the Kir channel has a third gate (called G-loop), which is located at the apex of the cytosolic domain and forms a girdle around the central fourfold axis (Pegan et al., 2005). Activation of the Kir channels depends on the signaling lipid phosphatidylinositol 4,5-bisphosphate (PIP2) (Shu and Hille, 2008). PIP2 binds at the interface between the pore-forming domain and the cytosolic domain in Kir2.2.
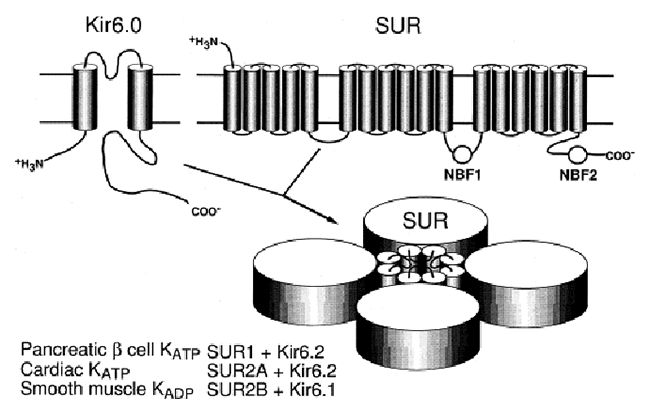
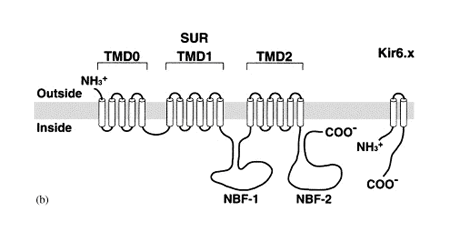
Acetylcholine activates K+ channels (KAch) in pacemaker SA and atrial cells. The acetylcholine activated current (IKAch) results from a tetrameric complex of four pore-forming subunits (Kir6.1 encoded by KCNJ3 gene and Kir6.2 encoded by KCNJ11 gene) and four regulatory sulfonylurea receptors (SUR1 and SUR2A/B encoded by the ABCC8 and ABCC9 gene, respectively) (Billman 2008). SUR2A subunit cause channel opening when bound to MgADP or channel openers (i.e., diazoxide). Following the binding of acetylcholine to muscarinic M2 receptors, the Gbg subunits of the PTX-sensitive G protein (Go/Gi family) activate the KAch channel by interacting directly to its cytoplasmatic N- and C-termini. Activation of IKAch hyperpolarizes the membrane potential, slows the spontaneous firing rate of the pacemaker cells of the SA and AV nodes and delays AV conduction (Tamargo et al., 2004). Recent data indicate that KATP channels in mice differ in a chamber-specific fashion: Kir6.1/SUR2B-containing channels are prominent in conduction system myocytes, atrial KATP channels are formed by Kir6.2/SUR1 and ventricular channels contain Kir6.2/SUR2A (Bao et al., 2011; Flagg et al., 2008).
3. FOUR TRANSMEMBRANE SEGMENTS AND TWO PORE (K2P) CHANNEL K2P channels are abundant in both excitable and non-excitable cells, where they play diverse functions (Enyedi and Czirjak, 2010). Structurally, each mammalian K2P channel has four TMs and two pore-forming domains. Thus, the biological assembly of a K2P channel is a dimer (Miller et al., 2012; Brohawn et al., 2014; Lolicato et al., 2014; Dong et al., 2016). They have intracellular N- and C-termini but functional K2P channels, unlike Kv and Kir channels that assemble as tetramers, exist as homodimers or heterodimers. Each of the (two) pore domains in each α-subunit contributes to the formation of the K+-selective pore (Lesage and Lazdunski 2000; Pael and Honore 2001; O´Connel et al., 2002). The conventional G(Y/F)G of K+ -selective motif is preserved in the first pore loop, but it is replaced by G(F/L)G in the second. K2P currents display little time- or voltage-dependence and I/V curves can be described by the Goldman-Hodgkin-Katz (GHK) equation, thus regulating resting membrane potential and excitability. There are four classes of cardiac K2P: TASK, TWIK, TREK and THIK.
Seino S, Miki T. Physiological and pathophysiological roles of ATPsensitive K+ channels. Prog Biophys Mol Biol 2003;81:133-76. Abbott GW and Goldstein SA. Disease-associated mutations in KCNE potassium channel subunits (MiRPs) reveal promiscuous disruption of multiple currents and conservation of mechanism. FASEB J 2002;16:390-400. Abbott GW, Butler MH, Bendahhou S, et al. MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell 2001104:217-231. Abbott GW, Goldstein S. Potassium channel subunits encoded by the KCNE gene family: physiology and pathophysiology of the minK-related peptides (MiRPs). Mol Interventions 2001;1: 95–107. Accili EA, Kiehn J, Yang Q, et al. Separable Kvbeta subunit domains alter expression and gating of potassium channels. J Biol Chem 1997;272:25824-25831. Ackerman MJ, Tester DJ, Porter C.J. Swimming, a gene-specific arrhythmogenic trigger for inherited long QT syndrome. Mayo Clin Proc. 1999;74:1088–1094. Alexander S, Mathie A, Peters J. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol 2011;164:S1-S364. An WF, Bowlby MR, Betty M, et al. Modulation of A-type potassium channels by a family of calcium sensors. Nature 2000;403:553-556. Anderson CL, Delisle BP, Anson BD, et al. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. Bao L, Kefaloyianni E, Lader J, Hong M, Morley G, Fishman GI, Sobie EA, Coetzee WA. Unique properties of the ATP-sensitive K⁺ channel in the mouse ventricular cardiac conduction system. Circ Arrhythm Electrophysiol 2011;4:926-935. Barhanin J, Lesage F, Guillemare E, et al. KvLQT1 and lsK (minK) proteins associate to form the IKs cardiac potassium current. Nature 1996;384:78-80. Ben-Abu Y, Zhou Y, Zilberberg N, et al. Inverse coupling in leak and voltage-activated K? channel gates underlies distinct roles in electrical signaling. Nat Struct Mol Biol 2009;16:71–79. Bezanilla F. The voltage sensor in voltage-dependent ion channels. Physiol Rev 2000; 80: 555-592. Bianchi L, Shen ZJ, Dennis AT, et al. Cellular dysfunction of LQT5-minK mutants: abnormalities of I-Ks, I-Kr and trafficking in long QT syndrome. Hum Mol Genet 1999;8:1499-1507. Billman GE. The cardiac sarcolemmal ATP-sensitive potassium channel as a novel target for anti-arrhythmic therapy. Pharmacol Ther 2008;120:54-70. Bosch RF, Zeng X, Grammer JB, Popovic K, Mewis C, Kuhlkamp V. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc Res 1999;44:121–131. Cabral JHM, Lee A, Cohen SL, et al. Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell 1998;95:649-656. Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010;67:915–928. Chen J, Zou A, Splawski I, et al. Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. J Biol Chem 1999;274:10113–10118. Chen QY, Zhang DM, Gingell RL, et al. Homozygous deletion in KVLQT1 associated with Jervell and Lange-Nielsen syndrome. Circulation 1999;99: 1344-1347. Chouabe C, Neyroud N, Guicheney P, et al. Properties of KvLQT1 K_ channel mutations in Romano-Ward and Jervell and Lange-Nielsen inherited cardiac arrhythmias. EMBO J 1997;16:5472-5479. Chouabe C, Neyroud N, Richard P, et al. Novel mutations in KvLQT1 that affect I-ks activation through interactions with Isk. Cardiovasc Res 2000;45:971-980. Clancy CE, Kass RS. Inherited and acquired vulnerability to ventricular arrhythmias: Cardiac Na+ and K+ Channels. Ohysiol Rev 2005;85:33-47. Coetzee W, Amarillo Y, Chiu J, et al. Molecular diversity of K+ channels. Ann N Y Acad Sci 1999;868:233-285. Cohen A, Ben-Abu Y, Zilberberg N. Gating the pore of potassium leak channels. Eur Biophys J. 2009;39:61-73. Cuello LG, Jogini V, Cortes DM, et al. Structural mechanism of C-type inactivation in K(?) channels. Nature 2010;466:203–208. Cui J, Melman Y, Palma E, et al. Cyclic AMP regulates the HERG K+ channel by dual pathways. Curr Biol 2000;10:671–674. Delisle BP, Underkofler HA, Moungey BM, et al. Small GTPase determinants for the Golgi processing and plasmalemmal expression of human ether-a-go-go related (hERG) K+ channels. J Biol Chem 2009;284:2844–2853. Delpón E, Valenzuela C, Pérez O, et al. Propafenone preferentially blocks the rapidly activating component of delayed rectifier K+ current in guinea-pig ventricular myocytes. Voltage-independent and time-dependent block of the slowly activating component. Circ. Res. 1995;76: 223-235. Donaldson MR, Jensen JL, Tristani-Firouzi M, et al. PIP2 binding residues of Kir2.1 are common targets of mutations causing Andersen syndrome. Neurology 2003;60:1811-1816. Dong YY, Pike AC, Mackenzie A, et al. K2P channel gating mechanisms revealed by structures of TREK-2 and a complex with Prozac. Science. 2015;347:1256–1259. Doyle DA, Morais-Cabral J, Pfuetzner RA, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 1998;280:69-74. Dvir M, Peretz A, Haitin Y. Recent molecular insights from mutated Iks channels in cardiac arrhythmia. Curr Opin Pharmacol. 2014;15:74–82. England SK, Uebele VN, Shear H, et al. Characterization of a voltage-gated K+ channel beta subunit expressed in human heart. Proc Natl Acad Sci USA 1995;92:6309-6313. Enyedi P, Czirják G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev 2010;90:559-605. Flagg TP, Kurata HT, Masia R, et al. Differential structure of atrial and ventricular KATP: atrial KATP channels require SUR1. Circ Res 2008;103:1458-65. Franqueza L, Lin M, Splawski I, et al. Long QT syndrome-associated mutations in the S4–S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J Biol Chem 1999;274:21063-21070. Gong Q, Zhang L, Vincent GM, et al. Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense-mediated mRNA decay in human long-QT syndrome. Circulation 2007;116:17–24. Grover GJ and Garlid KD. ATP-sensitive potassium channels: a review of their cardioprotective pharmacology. J Mol Cell Cardiol 2000;32:677-695. Gulbis JM, Zhou M, Mann S, et al. Structure of the cytoplasmic beta subunit-T1 assembly of voltage-dependent K+ channels. Science 2000;289:123-127. Hanlon MR, Wallace BA. Structure and function of voltage-dependent ion channel regulatory β subunits. Biochemistry 2002;41:2886– 94. Heginbotham L, Lu Z, Abramson R, MacKinnon R. Mutations in the K+ channel signature sequence. Biophys J 1994;66:1061-1067. Hibino H, Inanobe A, Furutani K, et al. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 2010;90:291-366. Hoorntje T, Alders M, van Tintelen P, et al. Homozygous premature truncation of the HERG protein: the human HERG knockout. Circulation 1999;100:1264–1267. Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 1990;250:533-538. Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron 1991;7:547-556. Huang CL, Feng S, and Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 1998;391:803-806. Imai S, Osawa M, Takeuchi K, et al. Structural basis underlying the dual gate properties of KcsA. Proc Natl Acad Sci USA 2010;107:6216–6221. Jenkinson DH. Potassium channels-multiplicity and challenges. Br J Pharmacol 2006; 147(Suppl.1): S63-S71. Jiang Y, Lee A, Chen J, et al. X-ray structure of a voltage-dependent K+ channel. Nature 2003;423:33–41. Johnson WH Jr, Yang P, Yang T, et al. Clinical, genetic, and biophysical characterization of a homozygous HERG mutation causing severe neonatal long QT syndrome. Pediatr Res 2003;53:744–748. Jurkiewicz NK, Sanguinetti MC. Rate-dependent prolongation of cardiac action potentials by a metanosulfonanilide class III antiarrhythmic agent. Specific block of rapidly activating delayed rectifier K+ current by dofetilide. Circ Res 1993;72:75-83. Kapplinger JD, Tester DJ, Salisbury BA, et al. Spectrum and prevalence of mutations from the first 2, 500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm 2009;6:1297–1303. Kass RS, Moss AJ. Long QT sndrome: novel insights into the mechanisms of cardiac arrhythmias. J Clin Invest 2003;112:810-815. Keating M, Atkinson D, Dunn C. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science. 1991;252:704–706. Kuang Q, Purhonen P, Hbert H. Structure of potassium channels. Cell. Mol. Life Sci. 2015;72:3677–3693. Kurokawa J, Abriel H, Kass RS. Molecular basis of delayed rectifier current I(Ks) in heart. J Mol Cell Cardiol. 2001;33:873–882. Kurokawa J, Chen L, and Kass RS. Requirement of subunit expression for cAMP-mediated regulation of a heart potassium channel. Proc Natl Acad Sci USA 2003;100: 2122–2127. Kuryshev YA, Gudz TI, Brown AM, et al. KChAP as a chaperone for specific K+ channels. Am J Physiol Cell Physiol 2000;278:C931-C941. Lees-Miller JP, Duan Y, Teng GQ, et al. Novel gain-of-function mechanism in K+ channel-related long-QT syndrome: altered gating and selectivity in the HERG1 N629D mutant. Circ Res 2000;86:507-513. Lesage F and Lazdunski M. Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol 2000;279:F793–F801. Liman ER, Hess P, Weaver F, et al. Voltage-sensing residues in the S4 region of a mammalian K+ channel. Nature 1991;353:752-756. Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science 2005;309:903–908. Lopatin AN, Nichols CG. Inward rectifiers in the heart: an update on IK1. J Mol Cell Cardiol 2001;33:625–638. López-Barneo J, Hoshi T, Heinemann SH, et al. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Recept Channels 1993;1:61-71. Lüscher C, Slesinger PA. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci 2010; 11: 301-315. MacKinnon R, Yellen G. Mutations affecting TEA blockade and ion permeation in voltage-activated K+ channels. Science 1990;250:276-279. MacKinnon R. Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature 1991;350:232-235. MacKinnon R. Potassium channels. FEBS Lett 2003;555:62-65. Martens JR, Kwak Y-G, Tamkun MM. Modulation of Kv channel α/β subunit interactions. Trends Cardiovasc Med 1999;9:253-258. Marx SO, Kurokawa J, Reiken S, et al. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 2002;295:496-499. McCrossan ZA, Abbott GW. The MinK-related peptides. Neuropharmacology 2004;47:787-821. McDonald TV, Yu Z, Ming Z, et al. A minK-HERG complex regulates the cardiac potassium current I(Kr). Nature 1997;388:289-292. Miller AN, Long SB. Crystal structure of the human two-pore domain potassium channel K2P1. Science. 2012;335:432–436. Mitcheson JS, Chen J, Sanguinetti MC. Trapping of a methanesulfonanilide by closure of the HERG potassium channel activation gate. J Gen Physiol 2000;115:229–240. Morais Cabral JH, Lee A, Cohen SL, et al. Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell 1998;95:649–655. Morais-Cabral JH, Zhou Y, MacKinnon R. Energetic optimization of ion co nduction rate by the K+ selectivity filter. Nature 2001;414:37–42. Morales MJ, Castellino RC, Crews AL, et al. A novel beta subunit increases rate of inactivation of specific voltage-gated potassium channel alpha subunits. J Biol Chem 1995;270:6272-6277. Moss AJ, Shimizu W, Wilde .A. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481–2489. Moss AJ, Zareba W, Kaufman ES, et al. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation 2002;105:794–799. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev 2005;85:1205-1253. O’Connell AD, Morton MJ, Hunter M. Two-pore domain K+ channels-molecular sensors. Biochim Biophys Acta 2002;1566:152 – 61. Ogielska EM, Zagotta WN, Hoshi T, et al. Cooperative subunit interactions in C-type inactivation of K channels. Biophys J 1995;69:2449-2457. Pandit SV, Berenfeld O, Anumonwo JM, et al. Ionic determinants of functional reentry in a 2-D model of human atrial cells during simulated chronic atrial fibrillation. Biophys J 2005;88:3806–3821. Papazian DM, Timpe LC, Jan YN, et al. Alteration of voltage-dependence of Shaker potassium channel by mutations in the S4 sequence. Nature 1991;349:305-310. Patel AJ, Honore´ E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci 2001;24:339–46. Patel SP, Campbell DL, Strauss HC. Elucidating KChIP effects on Kv4.3 inactivation and recovery kinetics with a minimal KChIP2 isoform. J Physiol 2002;545:5-11. Paulussen A, Raes A, Matthijs G, et al. A novel mutation (T65P) in the PAS domain of the human potassium channel HERG results in the long QT syndrome by trafficking deficiency. J Biol Chem 2002;277:48610–48616. Pegan S, Arrabit C, Zhou W, et al. Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci 2005;8:279–287. Piechotta PL, Rapedius M, Stansfeld PJ, et al. The pore structure and gating mechanism of K2P channels. EMBO J. 2011;30:3607–3619. Plaster NM, Tawil R, Tristani-Firouzi M, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s Syndrome. Cell 2001;105:511-519. Pongs O, Schwarz JR. Ancillary subunits associated with voltage-dependent K+ channels. Physiol Rev. 2010; 90: 755-796. Pongs O. Molecular biology of voltage-dependent potassium channels. Physiol Rev 1992;72:S69-S88. Pourrier M, Schram G, Nattel S. Properties, expression and potential roles of cardiac K+ channel accesory subunits: MinK, MiRPs, KChIP and KChAP. J Membr Biol 2003;194:141– 52. Rasmusson RL, Morales MJ, Wang S, et al. Inactivation of voltage-gated cardiac K+ channels. Circ Res 1998;82:739-750. Sanguinetti M, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature 2006;440:463-469. Sanguinetti MC, Jurkiewicz NK. Delayed rectifier outward K_ current is composed of two currents in guinea pig atrial cells. Am J Physiol Heart Circ Physiol 1991;260: H393–H399. Sanguinetti MC, Curran ME, Zou A, et al. Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature 1996;384:80-83. Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J Gen Physiol 1990;96:195-215. Sanguinetti MC, Xu QP. Mutations of the S4-S linker alter activation properties of HERG potassium channels expressed in Xenopus oocytes. J Physiol 1999;514:667-675. Sanguinetti MC. HERG1 channelopathies. Pflugers Arch. 2010;460:265-276. Sansom MS, Shrivastava IH, Bright JN, Tate J, Capener CE, Biggin PC. Potassium channels: structures, models, simulations. Biochim Biophys Acta. 2002;1565:294–307. Satler CA, Walsh EP, Vesely M, et al. Novel missense mutation in the cyclic nucleotide-binding domain of HERG causes long QT syndrome. Am J Med Genet 1996;65:27-35. Schram G, Pourrier M, Nattel S. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res 2002;90:939-950. Schwartz PJ, Priori SG, Spazzolini C. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. Seino S, Miki T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol 2003;81:133–76. Shushi L, Kerem B, Goldmit M, et al. Clinical, genetic, and electrophysiologic characteristics of a new PAS-domain HERG mutation (M124R) causing long QT syndrome. Ann Noninvasive Electrocardiol. 2005;10:334–341. Spector PS, Curran ME, Zou A, et al. Fast inactivation causes rectification of the IKr channel. J Gen Physiol 1996;107:611-619. Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 2000;102:1178-1185. Stengl M, Volders PGA, Thomsen MB, et al. Accumulation of slowly activating delayed rectifier potassium current (IKs) in canine ventricular myocytes. J Physiol 2003;551:777–786. Suh BC, Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys 2008;37:175–195. Tamargo J, Caballero R, Gómez R, et al. Pharmacology of cardiac potassium channels. Cardiovasc Res 2004;62:9-33. Tristani-Firouzi M, Sanguinetti MC. Structural determinants and biophysical properties of HERG and KCNQ1 channel gating. J Mol Cell Cardiol 2003;35:27-35. Tseng G-N. IKr: the hERG channel. J Mol Cell Cardiol 2001;33:835-849. Vandenberg CA. Inward rectification of a potassium channel in cardiac ventricular cells depends on internal magnesium ions. Proc Natl Acad Sci USA 1987;84:2560-2564. Voigt N, Trausch A, Knaut M, et al. Left-to-right atrial inward-rectifier potassium current gradients in patients with paroxysmal versus chronic atrial fibrillation. Circ Arrhythm Electrophysiol 2010;3:472–480. Volders P, Stengl M, van Opstal J, et al. Probing the contribution of IKs to canine ventricular repolarization: key role for beta-adrenergic receptor stimulation. Circulation 2003;107:2753-2760. Wang J, Trudeau MC, Zappia AM, Robertson GA. Regulation of deactivation by an amino terminal domain in Human Ether-a-go-go-related Gene potassium channels. J Gen Physiol 1998;112:637–647. Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 1996;12:17-23. Wei AD, Gutman GA, Aldrich R, et al., International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacological Reviews. 2005;57: 463–72. Wible BA, Yang Q, Kuryshev YA, et al. Cloning and expression of a novel K+ channel regulatory protein, KChAP. J Biol Chem 1998;273:11745-11751. Wu J, Ding WG, Horie M. Molecular pathogenesis of long QT syndrome type 1. J Arrhythm. 2016;32:381-388. Yellen G. The voltage-gated potassium channels and their relatives. Nature 2002;419:35-42. Yokoshiki H, Sunagawa M, Seki T, et al. ATP-sensitive K+ channels in pancreatic, cardiac, and vascular smooth muscle cells. Am J Physiol 1998;274:C25-37. Yu H, Wu J, Potapova I, et al. MinK-related peptide 1: a beta subunit for the HCN ion channel subunit family enhances expression and speeds activation. Circ Res 2001;88:E84-E87. Yuan P, Leonetti MD, Pico AR, et al. Structure of the human BK channel Ca2+-activation apparatus at 3.0 A resolution. Science. 2010;329:182-186. Zagotta WN, Aldrich RW. Voltage-dependent gating of Shaker A-type potassium channels in Drosophila muscle. J Gen Physiol 1990;95:29-60. Zeng J, Laurita KR, Rosenbaum DS, et al. Two components of the delayed rectifier K+ current in ventricular myocytes of the guinea pig type. Theoretical formulation and their role in repolarization. Circ Res 1995;77:140–152. Zhang M, Jiang M, Tseng GN. minK-related peptide 1 associates with Kv4.2 and modulates its gating function: potential role as beta subunit of cardiac transient outward channel? Circ Res 2001;88:1012-1019. Zhao JT, Hill AP, Varghese A, et al. Not All hERG pore domain mutations have a severe phenotype: G584S has an inactivation gating defect with mild phenotype compared to G572S, which has a dominant negative trafficking defect and a severe phenotype. J Cardiovasc Electrophysiol. 2009;20:923–930. Zhou W, Qian Y, Kunjilwar K, et al. Structural insights into the functional interaction of KChIP1 with Shal-type K+ channels. Neuron 2004;41:573-586. Zhou Y, Morais-Cabral JH, Kaufman A, et al. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature 2001;414:43–48. Zhou Z, Gong Q, Epstein ML, et al. HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. J Biol Chem 1998;273:21061–21066. Zou A, Curran ME, Keating MT, et al. Single HERG delayed rectifier K+ channels in Xenopus oocytes. Am J Physiol 1997;272:H1309–H1314. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
| Aviso legal Esta obra está bajo una licencia de Creative Commons Reconocimiento-NoComercial-SinObraDerivada 4.0 Internacional |
