|
TRPM4 channels Canalopatías Referencias
La caracterización de un mutante de la mosca Drosophila melanogaster que presentaba un defecto en la detección de luz y mostraba solo "potenciales transitorios inducidos por la luz" (TRP) en lugar de la respuesta mantenida normal llevó a la identificación de canales permeables al Ca2+ denominados canales de TRP (Cosens y Manning, 1969) . La gran familia de genes TRP humanos codifica canales catiónicos de potencial del receptor transitorio (TRP, Transient Receptor Potential) que forman canales catiónicos no selectivos que permiten la entrada Ca2+ y Na+ , aunque según la isoforma, la permeabilidad y selectividad para cationes mono o divalentes varía sustancialmente (de 100:1 a 0.05:1). En los mamíferos, se han identificado 28 genes del canal TRP en varias orgánulas de la mayoría de los tipos de células y tejidos (Owsianik et al., 2006; Nilius y Szallasi, 2014). En los mamíferos, se han identificado 28 genes del canal TRP que pueden dividirse en seis subfamilias (Owsianik et al., 2006; Nilius y Szallasi, 2014):
Varios miembros de la familia de TRP (TRPC1, TRPC3-7, TRPV1, TRPV2, TRPV4, TRPM2, TRPM4, TRPM6, TRPM7 y TRPP2) se expresan en el miocardio, en cardiomiocitos, fibroblastos, células endoteliales y células de músculo liso vascular (Alonso-Carbajo et al., 2017) Sin embargo, centraremos nuestra atención específicamente en el canal TRPM4.
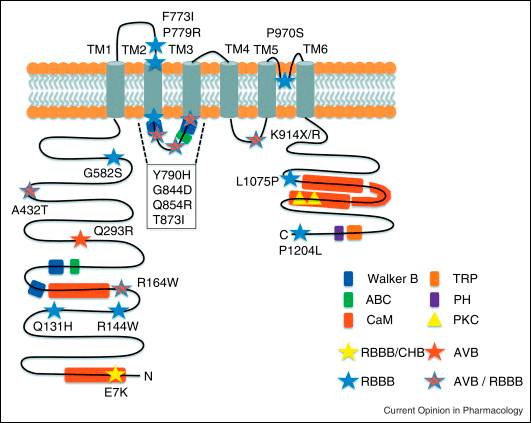
El gen TRPM4 se localiza en el cromosoma humano 19 y codifica una proteína de 1214 aminoácidos. Los canales de Melastatina 4 de potencial de receptor transitorio (TRPM4) presentan seis regiones de extensión transmembrana (S1-S6), un bucle de formación de poros entre S5-S6 y sus extremos C y terminales N son intracelulares. El canal funcional es un homotetramero y cada subunidad presenta sitios de unión para calmodulina, ATP y sitios de unión a fosfatidil inositol 4,5-bisfosfato (PIP2), motivos Walker B (es decir, motivos de unión a ATP o GTP que se encuentra en muchas proteínas de unión de nucleótidos), un sitio de glicosilación y sitios de fosforilación para la proteína quinasa A (PKA) y la proteína quinasa C (PKC) (Figura) (Kruse y Pongs, 2014; Guinamard et al., 2015).
Los canales de TRPM4 son impermeables al Ca2+, pero debido a su alta permeabilidad al Na+ es posible que a potenciales de reposo fisiológicos puedan modular la despolarización de la membrana y por lo tanto regular la función celular (Owsianik et al., 2006). A potenciales de membrana negativos, los canales de TRPM4 facilitan la entrada de Na+ en la célula, lo que despolariza de la membrana celular, mientras que a los potenciales de membrana positivos pueden facilitar el flujo de salida de K+, lo que facilita la repolarización de la membrana. De hecho, los miocitos auriculares aislados de ratones Trpm4−/− exhiben una DPA que es un 20% más corta que la de los PA de animales normales, lo que confirma que los canales TRPM4 están implicados en la morfología del PA auricular (Simard et al., 2013). En miocitos del nódulo sino-auricular, los canales de TRPM4 pueden desempeñar un papel en la despolarización diastólica espontánea y en la regulación del automatismo cardiaco (Hof et al., 2013) y contribuir a regular la repolarización auricular (Demion et al., 2014; Hof et al., 2013; Simard et al., 2013). Debido a que la [Ca2+] libre en el citoplasma durante la diástole es muy baja, es probable que el canal TRPM4 se encuentre en estado cerrado en reposo, pero podría activarse cuando la [Ca2+] libre aumenta durante las últimas fases de la repolarización. Por lo tanto, los canales TRPM4 son un candidato molecular para la corriente catiónica monovalente no selectiva activada con Ca2+ (NSCCa) descrita en cardiomiocitos de humanos (Mathar et al., 2014). Canalopatías. Se han identificado mutaciones en el gen TRPM4 en pacientes con bloqueo cardíaco familiar progresivo tipo I (PFHBI: Progressive familial heart block type I) y enfermedad de conducción cardíaca aislada (ICCD: solated cardiac conduction disease) que dan lugar a diversos fenotipos, incluidos bloqueo AV, bloqueo de la rama derecha, bradicardia y Síndrome de Brugada (Kruse y Pongs, 2014). Alonso-Carbajo L; Kecskes M, Jacobs G, et al. Muscling in on TRP channels in vascular smooth muscle cells and cardiomyocytes. Cell Calcium 2017;66:48-61.
Cosens DJ, Manning A. Abnormal electroretinogram from a Drosophila mutant. Nature 1969;224:285–287.
Demion M, Thireau J, Gueffier M, et al. Trpm4 gene invalidation leads to cardiac hypertrophy and electrophysiological alterations. PLoS ONE 2014;9:e115256.
Guinamard R, Bouvagnet P, Hof T, et al. TRPM4 in cardiac electrical activity. Cardiovasc Res. 2015;108:21-30.
Hof T, Simard C, Rouet R, et al. Implication of the TRPM4 nonselective cation channel in mammalian sinus rhythm. Heart Rhythm 2013;10:1683–1689.
International Union of Basic and Clinical Pharmacology TRP Channel Database (www.iuphar-db.org/index.jsp)
Kruse M, Pongs O. TRPM4 channels in the cardiovascular system, Curr.Opin. Pharmacol. 2014;15: 68–73.
Launay P, Fleig A, Perraud AL, et al. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 2002, 109:397-407
Liu H, Chatel S, Simard C, et al.: Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PLoS ONE 2013, 8:e54131.
Mathar I, Kecskes M, Van der Mieren G, et al. Increased beta-adrenergic inotropy in ventricular myocardium fromTrpm4-/- mice, Circ. Res. 2014;114:283–294.
Nilius B, Szallasi A. Transient receptor potential channels as drug targets: from the science of basic research to the art of medicine. Pharmacol Rev 2014;66:676-814.
Owsianik G, Talavera K, Voets T, Nilius B. Permeation and selectivity of TRP channels. Annu Rev Physiol 2006; 68: 685-717. |
|
|
| Aviso legal Esta obra está bajo una licencia de Creative Commons Reconocimiento-NoComercial-SinObraDerivada 4.0 Internacional |
