En individuos sanos el intervalo QT del ECG oscila entre 350 y 450 ms en los varones y entre 360 y 460 ms en las mujeres. El SQTC es una enfermedad arritmogénica primaria que se caracteriza por un intervalo QT del ECG anormalmente corto (QTc ≤ 320 ms) o de un intervalo QTc <360 ms y 1 o más de los siguientes hallazgos: historia de parada cardiaca o síncope, historia familiar de MSC antes de los 40 años de edad o antecedentes familiares de SQTC (Priori et al., 2015).
El SQTC se asocia con taquiarritmias auriculares y ventriculares, particularmente en reposo, y MSC (Bjerregaard et al., 2005; Patel et al., 2010). Sin embargo, existe controversia sobre el valor de corte exacto para definir un QT corto y dado que solo se ha identificado un número limitado de pacientes con SQTC, la prevalencia del síndrome es incierta, aunque parece que la aparición de una MSC como primera manifestación no es infrecuente.
Los datos de más de 10.000 adultos sugieren que, en la población sana, la prevalencia de QTc <340 ms es aproximadamente del 0.1% (Anttonen et al., 2007). Por lo tanto, los hombres con un QTc ≤330 ms y las mujeres con un QTc ≤340 ms tienen QT anormalmente corto se debe considerar que presentan un SQTC, incluso si son asintomáticos. Sin embargo, se han reportado individuos con QTc <320 ms que alcanzaron la edad adulta sin arritmias potencialmente mortales (Kobza et al., 2009).
La enfermedad parece ser altamente letal en todos los grupos de edad, incluidos los niños en sus primeros meses de vida, y la probabilidad de un primer paro cardiaco a la edad de 40 años es >40% (Gaita et al., 2003). La MSC se ha descrito incluso en el primer año de vida, lo que sugiere que el SQTC puede ser la causa de muerte en algunos bebés. Un tercio de los casos presentaron muerte súbita cardiaca como su primera manifestación clínica, y el 80% de los sujetos tenían antecedentes personales o familiares de SCD (Ferrero-Miliani et al., 2010).
La clínica es muy variable, desde pacientes que están asintomáticos y que se diagnostican por la historia familiar, hasta los pacientes que presentan palpitaciones, FA paroxística o permanente, síncope, arritmias ventriculares y MSC en pacientes sin cardiopatía estructural. Sin embargo, el síncope es menos frecuente que en otros síndromes arritmogénicos. Los datos disponibles sugieren que los pacientes corren riesgo a lo largo de su vida, con un pico entre la segunda y la cuarta décadas, posiblemente debido al pico en los niveles plasmáticos de testosterona, que puede causar un acortamiento de la QT, especialmente en adolescentes varones. Las circunstancias de la aparición de los síntomas son muy variables, y se han informado episodios de MSC durante o después de un ruido fuerte, en reposo, durante el ejercicio y durante las actividades diarias, lo que sugiere que el disparador del episodio arrítmico puede estar determinado por el tipo de mutación.
El diagnóstico es relativamente sencillo y puede hacerse sólo con el electrocardiograma y la historia clínica del paciente (Tabla).
Tabla 1.Criterios diagnósticos del SQTC (Priori et al., 2015)
|
Recomendaciones
|
Clase
|
Nivel |
| En presencia de un QTc ≤340 ms. |
I |
C |
Considerar un SQTC en presencia de un QTc ≤360 ms y 1 o más de los siguientes hallazgos:
- Una mutación patogémica confirmada
- Historia familar de SQTC
- Historia de MSC a una edad inferior a los 40 años
- Supervivientes de un episodio de TV/FV en ausencia de enfermedad cardiaca
|
IIa |
C |
Sin embargo, es posible que en un examen rutinario del ECG no se observa ninguna anomalía y que ésta puede observarse en un ECG posterior o en un estudio de Holter 24-48 horas. Se debe sospechar un SQTC en individuos jóvenes con un intervalo QT corto en ECG de 12 derivaciones con síntomas arrítmicos, síncope inexplicado o FA a una edad temprana, FV resucitada y cuando existen antecedentes familiares de muerte súbita y/o SQTC (Patel et al., 2010). El intervalo QT debe medirse preferiblemente cuando la frecuencia cardíaca es <80 bpm, porque todas las fórmulas corrigen en exceso los intervalos QTc a frecuencias cardíacas más altas, lo que puede conducir a un diagnóstico falso negativo. La realización de un Holter de 24-48 horas y un estudio electrofisiológico que permita conocer el valor del periodo refractario ventricular y evaluar la inducibilidad de la FV y/o de la FA ayudan a confirmar el diagnóstico. Sin embargo, la presencia de un intervalo QT corto en ausencia de síntomas o historia familiar no puede considerarse como un SQTC. Intervalos QT cortos (<360 ms en varones y <370 ms en mujeres) han sido descritos en pacientes con FV idiopática.
Tabla 2.Criterios diagnósticos (tomados de Gollob et al. 2011)
|
QTc, ms
|
Puntos
|
< 370 |
1 |
< 350 |
2 |
< 330 |
3 |
Intervalo entre punto J y la onda T < 120 ms |
1 |
| Historia clínica |
Historia de un paro cardíaco repentino |
2 |
VT o VF polimórficos documentados |
2 |
Síncope inexplicable |
1 |
Fibrilación auricular |
1 |
| Historia familiar |
Pariente de primer o segundo grado con alta probabilidad de SQTC |
2 |
Pariente de primer o segundo grado con MSC autopsia negativa |
1 |
Síndrome de muerte súbita infantil |
1 |
| Genotipo |
Genotipo positivo |
2 |
Mutations of undetermined significance in a culprit gene |
1 |
|
Alta probabilidad de SQTC: ≥4 puntos, probabilidad intermedia: 3 puntos, baja probabilidad: ≤2 puntos. Electrocardiograma: debe registrarse en ausencia de modificadores que puedan acortar el QT. El intervalo entre el punto J y el pico de la onda T se debe medir en la derivación precordial que presente la onda T de mayor amplitud. Historia clínica: los eventos deben ocurrir en ausencia de una etiología identificable, incluida una cardiopatía estructural. Los puntos solo se pueden recibir por 1 de paro cardíaco, TV polimórfica documentada o síncope inexplicable. Historial familiar: los puntos solo se pueden recibir una vez en esta sección. FV = fibrilación ventricular; TV = taquicardia ventricular.
|
En pacientes con SQTC el intervalo QTc es <300 ms, con un rango de 220-300 ms en pacientes con una frecuencia cardíaca entre 60 y 85 latidos por minuto (lpm). Este acortamiento del QTc puede ser intermitente y es menos evidente en el SQTC3 (~360 ms). El acortamiento del potencial de acción en el SQTS es heterogénea, siendo más marcado en el epicardio o endocardio, lo que da lugar a un aumento en la dispersión transmural de la repolarización (TDR).
También se observa una pérdida de la adaptación del intervalo QTc a las variaciones de la frecuencia cardíaca, con una disminución de la pendiente de la curva que relaciona intervalo QTc con la frecuencia cardíaca, lo que puede facilitar el diagnóstico del SQTC (Patel et al., 2010). Hasta un 50% de los pacientes exhiben ondas T puntiagudas y simétricas en derivaciones precordiales (como en una hiperpotasemia), que se inician inmediatamente después de la onda S, un segmento ST muy corto o prácticamente ausente y la onda T se origina de la onda S. En el SQTC tipo 3, las ondas T son asimétricas, con una rama ascendente menos pronunciada seguida de una rama descendente rápida. En muchos ECG hay un intervalo Tpico-Tfinal prolongado y el cociente Tpico-Tfinal/QT, una indicación de la dispersión transmural de la repolarización, aumentado (Gupta et al., 2008). La dispersión de la repolarización y de la refractariedad sirven como sustrato para reentrada ya que promueven la aparición de áreas de bloqueo unidireccional. El marcado acortamiento de la longitud de onda del circuitp (producto del período refractario y la velocidad de conducción) es un factor adicional que promueve el mantenimiento de la reentrada.
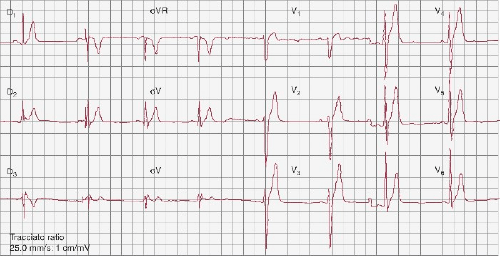
Figura. 1 ECG de un paciente con SQTC (QT/QTc, 300/300ms)(tomado de Monteforte et al., 2012).
Dado el reducido número de pacientes descritos con SQTC y que el intervalo QT varía con la frecuencia cardiaca, es necesario descartar otras causas que pueden cursar con un intervalo QT corto: taquicardia, hiperpotasemia, hipercalcemia, acidosis, intoxicación digitálica, hipertermia, aumento del tono parasimpático-vagal o niveles plasmáticos elevados de catecolaminas o testosterona. Un acortamiento paradógico del QT asociado a una disminución de la frecuencia cardiaca también debe ser tenido en cuenta.
En los pacientes con SQTC se produce un acortamiento no homogéneo, que es más evidente en las células endocárdicas y epicárdicas, que son las que presentan una duración potencial de acción más corta, lo que acentúa la dispersión de la repolarización entre éstas y las células M o de Purkinje. La heterogeneidad resultante de la repolarización y refractariedad interventricular e intraventricular generará un sustrato idóneo para el desarrollo de arritmias ventriculares malignas por reentrada asociadas con frecuencia a FA. De hecho, en pacientes con SQTC1 la TV se asocia con frecuencia a FA.
No se recomienda la utilización de estudios electrofisiológicos con estimulación ventricular programada para estratificar el riesgo de MSC (Priori et al., 2013).
Bases genéticas del SQTC
El SQTC es una enfermedad genética heterogénea, con un patrón de herencia autosómica dominante, pero sólo un 20% de los pacientes genotipados conocemos el gen responsable. Por tanto, el valor de las pruebas genéticas es limitado y no tiene implicaciones pronósticas, siendo necesarios nuevos estudios para identificar nuevos genes y establecer una correlación genotipo-fenotipo en el SQTC.
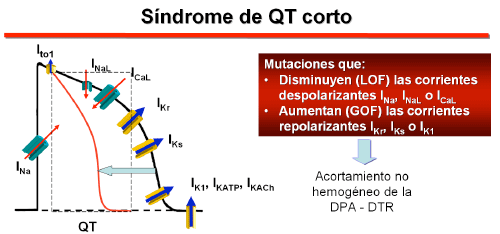
Hasta la fecha, mutaciones en 6 genes que codifican los canales de K+ y Ca2+ se han relacionado con el SQTC. El síndrome se asocia con mutaciones de ganancia de función en genes que codifican los canales de K+ (KCNH2, KCNQ1 y KCNJ2) o que producen una pérdida de función en los genes (CACNA1C, CACNB2B y CACNA2D1) que codifican diferentes subunidades del canal tipo L de Ca2+ (Gussak et al., 2000, Gaita et al., 2003, Bellocq et al., 2004, Brugada et al., 2004, Priori y otros, 2005, Antzelevitch et al., 2007). Una reducción de las corrientes repolarizantes y/o un aumento de las corrientes repolarizantes favorecen la rápida repolarización, un acortamiento del PA auricular y ventricular (intervalo QT) y la dispersión transmural de la repolarización, factores que representan un sustrato ideal para el desarrollo de mecanismos reentrantes, que en las aurículas pueden conducir a la FA y en los ventrículos a la TV/FV (Antzelevitch C, et al., 2007, Bjerregaard et al., 2005, Templin et al., 2011).
Las mutaciones en los canales de K+ conducen a un aumento de la corriente de salida durante la fase de meseta de la PA lo que acelera la repolarización ventricular y acorta el intervalo QT. La mutación con ganancia de función KCNH2 N588K localizada en la región de bucle S5-P en la boca externa del canal producía a una pérdida de rectificación de la IKr en el rango fisiológico de voltajes de la meseta del PA y desplazaba la curva de inactivación en +90 mV, lo que se traducía en un marcado aumento de la IKr durante la fase de meseta del PA y un marcado acortamiento de la duración del PA y del intervalo QT (Brugada et al., 2004). Curiosamente, esta mutación reducía la afinidad de los bloqueadores IKr, lo que tiene implicación directa en el tratamiento de SQT1. Los estudios electrofisiológicos invasivos revelaron que los periodos refractarios auricculares (PREA) son extremadamente cortos en SQTC (120-180 ms) y la vulnerabilidad auricular también aumenta, siendo frecuente la inducción de FA tras estimulación auricular programada. Utilizando un modelo experimental de SQTC1 que cursa con una ganancia de función de la IKr, Nof et al (2010) demostraron el desarrollo de una marcada dispersión de la repolarización y refractariedad entre la cresta terminal y el músculo pectinato en la aurícula derecha del perro. Un solo extrasístole introducido durante la ventana vulnerable inducía fácilmente FA. El sustrato reentrante era generado por el acortamiento del periro refractario y amplificado por la dispersión espacial de la repolarización intraauricular. La quinidina, pero no la lidocaína, prevenía la FA en este contexto.
Una mutación con cambio de sentido (V141M) en KCNQ1 se identificó en un neonato que presentaba bradicardia in utero e intervalos QT cortos y FA posparto (Hong et al., 2005). El modelado por ordenador demostró que la mutación acortaba la DPA de los miocitos ventriculares humanos y abolía la actividad del marcapasos del nódulo sinoauricular. La mutación KCNQ1 V307L producía un desplazamiento de -20 mV en la dependencia del voltaje de la activación de IKs y aceleraba la cinética de activación dando como resultado una ganancia de función (Bellocq et al., 2004). Curiosamente, esta mutación también se ha descrito en un paciente con FA (Chen et al., 2003). Otra mutación con ganancia de función (D172N) en el gen KCNJ2 que codifica el canal Kir2.1 que presenta rectificadora interna aumentaba la amplitud de la IK1 a potenciales entre -75 y -45 mV y desplazaba la corriente desde -75 mV hasta - 65 mV (Priori et al., 2005). Como resultado, la repolarización ventricular se aceleraba y el APD se acortaba marcadamente.
The SQTS1 is caused by mutations in KCNH2 (HERG). La missense mutación KCNH2 N588K suprime la rectificación de la IKr en el rango de voltajes fisiológicos y desplaza la inactivación voltaje-dependiente en 90 mV, lo que produce una marcada ganancia de función de la IKr durante la fase de meseta del PA y un marcado acortamiento del PA and of ventricular ERP, leading to AF y ventricular fibrillation (VF) (Brugada et al., 2004). Esta mutación, además, reduce la afinidad de los bloqueadores de la IKr, lo cual tiene implicación directa en el tratamiento de la SQTC1. La mutación KCNQ1 V307L produce un desplazamiento de -20 mV en la curva de activación de la IKs y acelera la cinética de activación que resulta en aumento de función (Bellocq et al., 2004). Curiosamente, esta mutación también se ha descrito en un paciente con FA (Chen et al., 2003). Otra mutación (D172N) en el gen KCNJ2 aumenta la IK1 a potenciales comprendidos entre -75 y -45 mV y el pico de corriente se desplaza desde -75 mV a - 65 mV (Priori et al, 2005). Como resultado la repolarización se acelera y se acorta la DPA ventricular.
Una reducción de la ICaL también puede acortar el intervalo QT. Se han descrito mutaciones de pérdida de función en el gen CACNA2D1 (Ser755Thr), que codifica para la subunidad auxiliar de Cavα2δ-1 del canal de Ca2+ tipo-L que está implicada en el tráfico del canal hacia la membrana y en la modulación de las propiedades biofísicas de la subunidad α1 en pacientes con SQT6 (Temlin et al., 2011). Mutaciones en CACNB2b (S481L) y CACNA1C (A39V y G490R) también causan una pérdida importante de función en la actividad del canal de calcio tipo-L (Antzelevitch et al., 2007). Algunas de estas mutaciones producen también una elevación del segmento ST en derivaciones precordiales que rememoran un SBr que puede ser el fenotipo dominante en estos pacientes.
Tabla 3.Genes que codifican canales iónicos cardiacos implicados en la génesis del SQTC
|
Síndrome
|
Locus
|
Gen
|
Proteína
|
Corriente
|
Función
|
SQTS1 (18-33%) |
7q35-q36 |
KCNH2 |
Subunidad α |
IKr |
(+) |
SQTS2 (<5%) |
11p15.5 |
KCNQ1 |
Kv7.1 α |
IKs |
(+) |
SQTS3 (<5%) |
17q23.1-24.2 |
KCNJ2 |
Kir2.1 α |
IK1 |
(+) |
SQT4 |
12p13.3 |
CACNA1C |
Subunidad α Cav1.2 |
ICa, L |
(-) |
SQT5 |
10p12 |
CACNB2B |
Subunidad β2 Cav1.2 |
ICa, L |
|
SQT6 |
7q21-q22 |
CACNA2D1 |
Subunidad α2/δ1 Cav1.2 |
ICa, L |
|
SQT7 |
3q22 |
SCN5A |
Subunidad α |
INa |
(-) |
SQT8 |
2q35 |
SLC4A3 |
Anion exchanger AE3 |
|
(-) |
SQT9 |
3p22.2 |
SCN10A |
INa (noncanonical α) |
INa |
(-) |
Tratamiento
La estrategia óptima para la prevención primaria de la parada cardiaca en pacientes con SQTC es incierta (Mazzanti et al., 2014). No disponemos de datos clínicos para cuantificar el riesgo de eventos arrítmicos durante la actividad física competitiva en pacientes SQTS.
En la actualidad, no existe una terapia farmacológica para prevenir las arritmias potencialmente mortales, pero debido a que la tasa de recurrencia de paro cardíaco se ha estimado en un 10% anual, se recomienda implantar un DAI en pacientes con diagnóstico de SQTS que (a) sean supervivientes de un paro cardiaco abortado, y/o (b) presentan TV sostenidas espontáneas, con o sin síncope (Recomendación I, C) (Priori et al., 2015). La decisión de insertar un DAI debe basarse en criterios clínicos (intervalo QT corto en el ECG junto con síntomas arrítmicos y antecedentes familiares sólidos de MSC) en lugar de genéticos o electrofisiológicos hasta que dispongamos de otras pautas para la estratificación del riesgo (Patel et al., 2010). Un problema único con los DAI en SQTC es que las ondas T altas y picudas que siguen de cerca a la onda R pueden interpretarse a como un intervalo R-R corto, provocando un choque inapropiado del DAI (Schrimpf et al., 2003). Villafane et al. informaron una tasa notablemente alta de terapias de CDI inapropiadas de hasta el 64% en seis años en una gran cohorte pediátrica de pacientes con SQTS. Además, la alta prevalencia de FA en pacientes con SQTS puede presentar un desafío clínico para la programación apropiada de ICD.
La información sobre la terapia farmacológica para SQTS es bastante limitada, y la mayoría de los datos disponibles pertenecen a SQTC1. El tratamiento farmacológico puede ser útil como complemento del DAI o puede utilizarse para la prevención primaria cuando el paciente rechaza un DAI o en niños pequeños en los que la implantación de un DAI puede ser problemática.
DAI: desfibrilador automático implantable; EEF: estudio electrofisiológico; EVP: estimulación ventricular programada; MSC: muerte súbita cardiaca; SQTC: síndrome del QT corto.
Estratificación de riesgos y tratamiento en el síndrome del QT corto (Priori et al., 2015)
|
Recomendaciones
|
Clase
|
Nivel
|
Se recomienda implantar un DAI para pacientes con diagnóstico de SQTC:
- Supervivientes a una parada cardiaca abortada
- Con TV sostenida espontánea documentadaHave documented spontaneous sustained VT
|
I |
C |
Se puede considerar quinidina o sotalol para pacientes con diagnóstico de SQTC cualificados para DAI pero que lo tienen contraindicado o lo rechazan
|
IIb |
C |
Se puede considerar quinidina o sotalol para pacientes asintomáticos con diagnóstico de SQTC e historia familiar de MSC
|
IIb |
C |
No se recomienda el EEF invasivo con EVP para la estratificación de riesgo de MSC
|
III |
C |
El tratamiento farmacológico puede ser útil como complemento del DAI o puede usarse para la prevención primaria cuando el paciente rechaza un DAI o en niños pequeños en los que la implantación de un DAI puede ser problemática. Gaita et al (2004) estudiaron los efectos de flecainida, sotalol, ibutilida e hidroquinidina en seis pacientes con SQT1. Solo la hidroquinidina prolongaba el intervalo QT a valores normales, aumentaba la refracteriedad ventricular, restablecía la relación QT-RR hacia la normalidad, disminuía la amplitud de la onda T del ECG y conertía la FV no inducible. En 53 pacientes del registro europeo de SQTC (QT medio 313 ms, 89% con antecedentes de paro cardíaco), la hidroquinidina normalizaba el intervalo QT y la refractariedad ventricular y prevenía la taquiarritmia ventricular durante el estudio electrofisiológico y los eventos arrítmicos durante un período de seguimiento de 64 meses. Los pacientes con mutaciones HERG (N588K, T618I) presentaban una mayor prolongación del QTc tras el tratamiento con hidroquinidina. Tras 2 años de seguimiento, la hidroquinidina era efectiva para prevenir la inducción de las taquiarritmias ventriculares y los eventos arrítmicos (Giustetto et al., 2011).
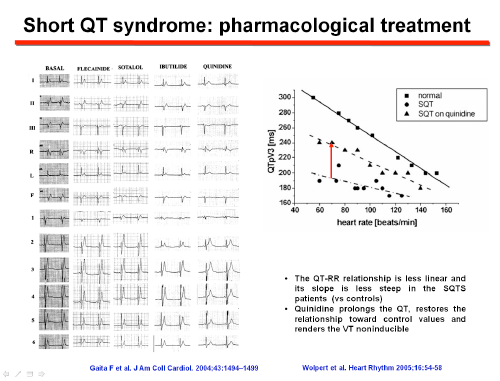
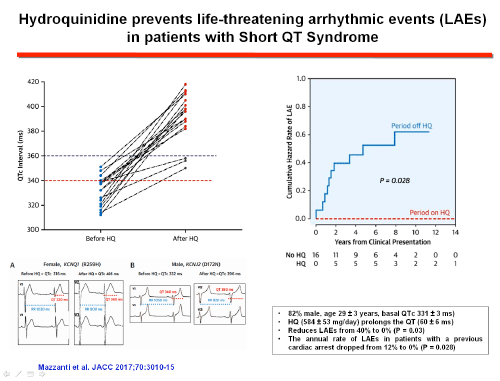
La eficacia de quinidina e hidroquinidina y el fracaso de los fármacos antiarrítmicos de clase IC y III (sotalol, amiodarona) en pacientes con la mutación KCNH2 N588K pueden relacionarse con las diferencias en su interacción con el canal HERG. La mayoría de los fármacos que prolongan el QT presentan una mayor afinidad por el estado inactivado de los canales HERG, lo que explica su eficacia reducida cuando una mutación elimina o reduce la inactivación del canal (Gaita et al., 2004; Perrin et al., 2008). Sin embargo, quinidina y hidroquinidina presentan una afinidad similar por los estados abiertos e inactivados del canal y es una terapia eficaz en pacientes con SQTC, restableciendo la relación QT-RR hacia el rango normal. La quinidina reduce la tasa de eventos de aproximadamente un 4.9% a un 0% anual, aunque los datos actuales se derivan de cohortes muy pequeñas con una duración de seguimiento moderada (Patel et al., 2010). Si bien su eficacia se ha demostrado en pacientes con SQTC1, la quinidina bloquea otras corrientes de K+ (Ito, IK1, IKr, y IKs), por lo que también podría ser eficaz en otras formas de SQTC. Schimpf et al (2007) demostraron que la disopiramida también aumentaba el intervalo QT y el período refractario ventricular en pacientes con SQTc1. Los pacientes que toman quinidina, hidroquinidina o dispiramida deben ser monitoreados cuidadosamente para detectar la prolongación del intervalo QT y posibles eventos pro-arrítmicos.
La quinidina o el sotalol pueden considerarse en pacientes con diagnóstico de SQTC que califiquen para un DAI pero que presenten una contraindicación para su implantación o cuando el paciente lo rechaza (Recomendación IIb, C). Igualmente, quinidina o sotalol pueden utilizarse en pacientes asintomáticos con diagnóstico de SQTC y antecedentes familiares de MSC (Recomendación IIb, C) (Priori et al., 2015).
A diferencia de SQTC1, los AAD de clase III podrían ser útiles en pacientes con SQTC2 y SQTC3 y sotalol, amiodarona y el nifekalant en SQTC con genotipo desconocido y se espera que sean clínicamente útiles en SQTC2 y SQTC3; sin embargo, disponemos de muy pocos estudios que hayan analizado la seguridad y eficacia de estos fármacos.
La FA es un problema clínico común en pacientes con SQTC. La propafenona ha demostrado ser eficaz para prevenir los episodios frecuentes de FA sin reaparición de arritmia durante más de dos años pero sin ningún efecto sobre el intervalo QT (Bjerregaard y Gussak, 2005). La flecainida prolonga el período refractario, pero no la inducción de VF en la estimulación programada.

Referencias
Anttonen O, Junttila MJ, Rissanen H, et al. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation 2007;116:714–20.
Antzelevitch C, Pollevick GD, Cordeiro JM et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–9.
Bjerregaard P, Gussak I. Short QT syndrome: mechanisms, diagnosis and treatment. Nat Clin Pract Cardiovasc Med. 2005;2:84–87.
Bjerregaard P, Gussak I. Short QT syndrome. Ann Noninvasive Electrocardiol 2005;10:436–40.
Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation.2004;109:2394-7.
Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004; 109: 30–35.
Chen YH, Xu SJ, Bendahhou S, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–254.
Ferrero-Miliani L, Holst AG, Pehrson S, et al. Strategy for clinical evaluation and screening of sudden cardiac death relatives. Fundam Clin Pharmacol. 2010;24:619–35.
Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: a familial cause of sudden death. Circulation 2003;108:965–70.
Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment, J Am Coll Cardiol 2004;43:1494–9.
Giustetto C, Schimpf R, Mazzanti A, et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol 2011;58:587–595.
Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol. 2011;57:802-12
Gupta P, Patel C, Patel H, et al. Tp-e/QT ratio as an index of arrhythmogenesis. J Electrocardiol 2008;41:567–574.
<p class="textoreferencias">Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome?. Cardiology. 2000;94:99-102.
Hong K, Piper DR, Diaz-Valdecantos A, et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero, Cardiovasc Res 2005;68:433–40.
Kobza R, Roos M, Niggli B, et al. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm 2009;6:652–7.
Mazzanti A, Kanthan A, Monteforte N, et al. Novel insight into the natural history of short QT syndrome. J Am Coll Cardiol 2014;63:1300–1308.
Monteforte N, Napolitano C, Napolitano C, et al. Genetics and arrhythmias: diagnostic and prognostic applications. Rev Esp Cardiol 2012;65:278-285.
Nof E, Burashnikov A, Antzelevitch C. Cellular basis for atrial fibrillation in an experimental model of short QT1: implications for a pharmacological approach to therapy. Heart Rhythm. 2010;7:251-7.
Patel C, Yan G-Z, Antzelevitch C. Short QT Syndrome: From bench to bedside. Circ Arrhythm Electrophysiol. 2010;3:401-408.
Perrin MJ, Kuchel PW, Campbell TJ, et al. Drug binding to the inactivated state is necessary but not sufficient for high-affinity binding to human ether-à-go-go-related gene channels. Mol Pharmacol. 2008;74:1443-52.
Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005; 96: 800–807.
Priori SG, Wilde AA, Horie M, et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm 2013;10:e85–e108.
Priori SG, Blomström-Lundqvist C, Mazzanti A, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J 2015;36:2793–2867.
Schimpf R, Antzelevitch C, Hsu LF, et al. The QT-interval in patients with a Brugada syndrome: is a shortening of the QT-time an existing and relevant ECG-pattern? Heart Rhythm. 2007;4:S188.
Schimpf R, Veltmann C, Giustetto C, et al. In vivo effects of mutant HERG K+ channel inhibition by disopyramide in patients with a short QT-1 syndrome: a pilot study. J Cardiovasc Electrophysiol 2007;18:1157–60.
Schimpf R, Wolpert C, Bianchi F, et al. Congenital short QT syndrome and implantable cardioverter defibrillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol, 2003;14:1273–7.
Templin C, Ghadri JR, Rougier JS et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6) Eur Heart J. 2011;32:1077–88.
