Síndrome de Brugada (SBr)
|
Es un síndrome clínico-electrocardiográfico, de transmisión autosómica dominante, penetrancia incompleta, con predominio en varones, caracterizado por una elevación persistente del segmento ST (≥ 2 mm) en las derivaciones precordiales derechas (V1-V3), con o sin bloqueo completo de rama derecha, que aparece en pacientes sin cardiopatía estructural (Brugada et al., 1992, 2002; Antzelevitch et al., 2005). Aún cuando se describió originalmente como una canalopatía en la que no hay defectos estructurales cardíacos, diferentes estudios han señalado anomalías cardíacas estructurales microscópicas en los corazones de pacientes con SBr que conducen a un enlentecimiento de la conducción cardíaca.
Su prevalencia se estima en 1-5/10.000 habitantes, aunque posiblemente sea muy superior, ya que muchos pacientes están asintomáticos (Mizusawa y Wilde, 2012). El SBr tipo 1 se presenta con mayor frecuencia en varones de origen asiático (japoneses 0.15–0.27%, filipinos 0.18%, japoneses de los Estados Unidos 0.15%) que en europeos (0%–0.017%) o norteamericanos (0.005–0.1%) (Antzelevitch et al., 2005, 2016). De hecho, en los varones jóvenes con corazones normales de algunos países del sudeste asiático el SBr es la segunda causa de muerte, sólo superada por los accidentes de tráfico (Veerakul y Nademanee, 2012). Sin embargo, desconocemos cómo el género modula la manifestación de la enfermedad (Wilde et al, 2002;.. Antzelevitch et al, 2005). En individuos pre-púberes no hay diferencias significativas de género en los valores del intervalo ST en las derivaciones V2 y V5, pero sí aparecen tras la pubertad en los varones (Ezaki et al., 2010). La terapia de deprivación de andrógenos reduce significativamente los niveles del ST en V2 y V5 en los varones, que ahora se asemejan a los de las mujeres de la misma edad, lo que sugiere que la testosterona modularía las corrientes iónicas que determinan la fase 1 temprana de la repolarización epicárdica ventricular. El efecto de los estrógenos (que reducen la amplitud de la Ito e inhiben el tráfico de los canales Kv4.3 y su expresión en la membrana) y las diferencias en la expresión y la densidad de la Ito entre ambos sexos (la densidad de la Ito es mayor en el epicardio en varones que en mujeres) también puede explicar el predominio de fenotipo del SBr en los varones (Di Diego et al., 2002).
La BrS es un trastorno genético autosómico dominante con una penetrancia incompleta ya menudo baja y un predominio masculino importante. Una hipótesis atractiva para explicar la penetrancia incompleta del SBr es la existencia de modificadores genéticos que pueden ser variantes comunes en SCN5A u otros genes. El SBr es genéticamente heterogéneo, asociado a mutaciones en diversos genes que conducen a: 1) una pérdida de función de las corrientes despolarizantes INa e ICa, y 2) una ganancia de función de las corrientes repolarizantes Ito o IKATP y 3) una reducción de la If (Juang y Horie, 2016) (Tabla). Por tanto, el SBr es el resultado de un desequilibrio entre corrientes que favorecen la repolarización sobre la despolarización.
En conclusión, se han identificado más de 25 genes de susceptibilidad en pacientes con SBr y es probable que la heterogeneidad genética sea aún mayor, ya que la detección de mutaciones en los genes conocidos permite identificar una mutación en tan solo ~ 25–30% de los pacientes clínicamente afectados, lo que indica que 65–70 % de pacientes con BrS permanecen sin resolver genéticamente. Como consecuencia, el valor de las pruebas genéticas para fines de diagnóstico es limitado y no hay evidencia de que los resultados de éstas influyan en el manejo clínico o la estratificación del riesgo en BrS (Priori et al., 2012). Debido a la baja prevalencia de mutaciones no relacionadas con el gen SCN5A, se ha sugerido que es razonable realizar inicialmente en la mayoría de los pacientes un estudio genético para detectar mutaciones de SCN5A, realizando el estudio adicional de otros genes menores solo en circunstancias especiales y no se recomienda realizar tests genéticas en el ausencia de un ECG de diagnóstico (Ackerman et al., 2011). Además, la interpretación cuidadosa de las variantes identificadas es extremadamente importante para los pacientes con SBr o los miembros de la familia, especialmente para los probandos individuales o para las familias pequeñas. Algunos de los genes identificados solo han magnificado los problemas de interpretación al incrementar el "ruido genético" de fondo. Por tanto, es muy importante los resultados de las pruebas genéticas como estrictamente probabilísticos, en lugar de binarios/deterministas, por naturaleza. Una gran cantidad de variantes permanecen en el "purgatorio genético", y este número seguirá aumentando según la secuenciación del genoma/exoma se utilicen con mayor frecuencia. Se debe tener cuidado al interpretar las variaciones genéticas en estos otros genes menores porque las variantes de codificación raras se observan en una medida similar tanto en los casos como en los controles.
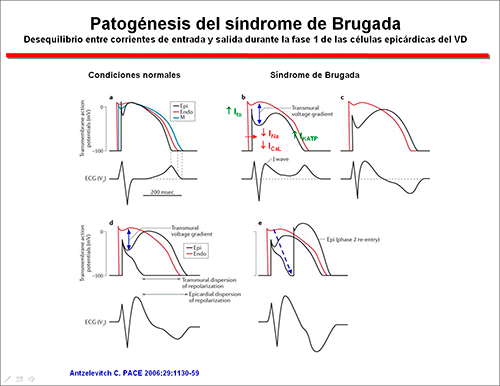
Las bases del SBr son desconocidas, existiendo un debate acerca de si en el SBr existe: 1) un trastorno de la repolarización asociado a un aumento en la dispersión transmural en la morfología del PA del ventrículo derecho que conduce a una pérdida de la morfología pico-cúpula a nivel del epicardio del ventrículo derecho que crearía la base para la aparición de arritmias por reentrada. 2) Un trastorno de la despolarización y de la velocidad de conducción en el ventrículo derecho (VD). Si estas dos hipótesis se excluyen mutuamente o si todos los casos de BrS se originan por el mismo mecanismo fisiopatológico, aún no está claro.
Figura 1 Una explicación alternativa del patrón electrocardiográfico del SBr es la hipótesis de despolarización que sugiere que existe un retraso de la conducción en el RVOT secundario a anomalías estructurales (p. ej., inflamación, hipertrofia, vacuolación, apoptosis de cardiomiocitos, infiltración fibrofatty y fibrosis) y una menor expresión de la connexina (Cx) 43 que crea las condiciones óptimas para la reentrada (Nagase et al., 2002, Postema et al 2008; Wilde et al., 2010; Elizari et al., 2007). Se ha observado un retraso significativo en la conducción regional, una reducción en el gradiente de activación, la presencia de electrogramas fraccionados y formación de líneas de bloqueo de conducción funcional en la pared libre anterolateral del RVOT en comparación con el resto y el apex del VD en los pacientes con SBr (Lambiase et al., 2009; Coronel et al., 2005; Postema et al., 2010). Además, Nademanedee et al (2011) encontraron en pacientes con un patrón de Brugada tipo 1 y episodios de TV/FV la presencia de potenciales tardíos de baja amplitud, duración prolongada y fraccionados que se localizaban exclusivamente en la pared anterior del epicardio RVOT. Estos resultados confirman la presencia de un retraso de la despolarización a este nivel que facilitaría el desarrollo de los circuitos de reentrada epicárdica que se agravaría con la reducción del INa. El retraso en la conducción en el RVOT produce un gradiente eléctrico desde el RV más positivo al RVOT, lo que lleva a una elevación del ST en las derivaciones precordiales derechas y, a medida que el RVOT se despolariza más tarde (durante la repolarización del RV), este gradiente se invierte y la corriente neta fluye desde el RVOT hacia el RV, lo que resulta en una onda T negativa en las mismas derivaciones precordiales derechas (Meregalli et al., 2005). Este mecanismo ha ganado apoyo al observarse que la ablación epicárdica del RVOT suprime el fenotipo del SBr. Sin embargo, el comportamiento típico en los pacientes con SBr al incrementar la frecuencia de estimulación era una disminución de la elevación del segmento ST, una respuesta opuesta a la esperada en presencia de una conducción discontinua que sugería que el deterioro de la conducción no era la causa principal del ECG de BrS (Zhang et al., 2015). De hecho, estos autores demostraron que ambos mecanismos, conducción lenta discontínua y dispersión de la repolarización estabn presentes en el RVOT de los pacientes con SBr y que el retraso de la conducción no se limitaba exclusivamente al RVOT. Por lo tanto, es posible que algunos de estos cambios puedan ser el resultado, en lugar de la causa del sustrato BrS, que puede crear un estado similar a la hibernación debido a la pérdida de contractilidad en el RVOT secundario a la pérdida del domo AP. Se ha propuesto que los cambios iónicos responsables de SBr (es decir, la pérdida de la función INa e ICa y la ganancia de función de la Ito) pueden alterar la morfología AP para reducir la seguridad de la conducción en las uniones de alta resistencia, como las regiones donde existe una fibrosis extensa (Hoogendijk et al., 2010).
Son muy variables, desde pacientes asintomáticos a aquellos en los que la primera manifestación es una SCD (Antzelevitch et al., 2005,2006). Las manifestaciones clínicas más comunes son el síncope y las convulsiones, respiración agónica o la MSC producida por episodios de FV no sostenida que ocurren por lo general durante el sueño o en reposo. La función sinusal es normal, aunque taquicardias supraventriculares, en particular, la fibrilación auricular (FA), están presentes en el 20-30% de los pacientes y, de hecho, la FA puede ser la primera manifestación de un SBr (Kusano et al., 2008). El bloqueo AV y los retrasos de conducción intraventricular (intervalo HV de 60-75 ms) también son parte del fenotipo del SBr (Smits et al., 2002) y un alto porcentaje de los pacientes presentan una TV (o FV) inducible durante la estimulación ventricular programada. En los estudios electrofisiológicos se observa con frecuencia bloqueos AV y retrasos de la conducción intraventricular (intervalo H-V 60-75 ms), que explica la ligera prolongación del intervalo PR y la morfología de bloqueo de rama derecha y hemibloqueo anterior izquierdo en el ECG. Sin embargo, los parámetros hemodinámicos son normales.
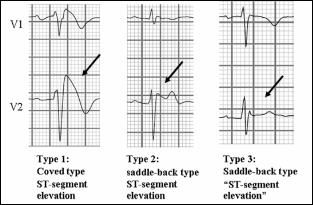
El ECG es muy variable a lo largo del tiempo incluso en un mismo paciente e incluso puede ser transitoriamente normal. Están los pacientes asintomáticos en quienes el electrocardiograma típico del síndrome se encuentra por casualidad durante un examen rutinario o durante un estudio a causa de la MSC de un familiar con SBr. En el otro extremo están los pacientes sintomáticos que han sido diagnosticados de síncope de causa desconocida o síncope vaso-vagal o de una FV idiopática, o aquellos en los que la administración de fármacos antiarrítmicos del grupo I desenmascara un patrón ECG oculto (Figura 1).
Figura 2
Es posible que los patrones electrocardiográficos difieran también según la mutación y que las variaciones observadas con el tiempo estén relacionados con cambios en el tono autonómico, la temperatura corporal o la frecuencia cardiaca (Benito et al, 2008; Mizusawa et al, 2012). La estimulación adrenérgica (isoproterenol), el ejercicio y el aumento de la frecuencia cardíaca disminuyen la elevación del segmento ST; de hecho, algunos pacientes con "tormentas de FV" pueden ser tratados eficazmente con una infusión i.v. de isoproterenol (Tanaka et al., 2001). Por el contrario, el ECG de Brugada a menudo es oculto y puede desenmascararse por fármacos y maniobras que aumenten el tono vagal, agonistas -adrenérgicos, bloqueantes β-adrenérgicos, fármacos antiarrítmicos del grupo I, antidepresivos tricíclicos o tetracíclicos, hiperpotasemia, hipopotasemia, hipercalcemia, alcohol, cocaína, comidas pesadas, estados febriles, bradicardia (Brugada et al., 2000; Veerakul y Nademanee, 2012). La lista de los fármacos que pueden desenmascarar el ECG de Brugada y que se deben evitar los pacientes con SBr se puede consultar en www. brugadadrugs.org (see Table).
La muerte súbita en pacientes con BrS generalmente ocurre durante el sueño o durante un bajo nivel de actividad física, lo que sugiere que las variaciones de la frecuencia cardíaca, el equilibrio simprovagagal, las hormonas y otros factores metabólicos pueden contribuir a este patrón circadiano. Es plausible que por la noche, durante el sueño, cuando el tono vagal suele aumentar y disminuye la actividad simpática, el sustrato es más susceptible a la arritmogénesis. Sin embargo, se necesitan más estudios para definir y comprender claramente la compleja interacción entre el sistema nervioso autónomo y los mecanismos arrítmicos de la EB. Según las guías de 2015 de la Sociedad Española de Cardiología para el tratamiento de pacientes con arritmias ventriculares y la prevención de la SCD (Priori et al., 2015), el diagnóstico del SBr se basa en la historia clínica y en la identificación del patrón tipo 1 en el ECG (sin o tras la administración de un bloqueantes de los canales de Na+) y la presencia de uno de los siguientes criterios: 1) historia de episodios de TV/FV espontáneos o de MSC abortada; 2) historia familiar de MSC; 3) una historia familiar de un patrón tipo 1 en el ECG y 4) la presencia de síntomas relacionados con TV/FV: síncope, convulsiones o respiración agónica durante el sueño. En cualquier caso, es necesario descartar otras enfermedades que producen una elevación del segmento ST del ECG (isquemia, miocarditis, hipercalemia, hipercalcemia, displasia ventricular arritmogénica o embolismo pulmonar.
Cuando el ECG es menos característico y en pacientes con un ECG normal es posible desenmascarar la elevación característica del segmento ST (patrón tipo I) en las derivaciones precordiales derechas tras la administración i.v. de fármacos que bloquean los canales de Na+ (ajmaline, flecainida, pilsicainida, procainamida) (Wilde et al., 2002; Atzelevitch et al., 2016). Estos fármacos producen un retraso adicional de conducción en el TSVD e inducen un gradiente transmural al acortar el PA más en las células epicárdicas que en las endocárdicas, lo que contribuye a la aparición de un patrón de tipo 1 en el ECG (Maregalli et al., 2005). Esta es la razón por la que se recomienda realiar un test de ajmalina en todos los pacientes con síncope de causa inexplicada o FV idiopática para excluir la presencia de un SBr. La infusión de estos fármacos debe terminar si el ensanchamiento del complejo QRS es ≥130% sobre la basal.
Aunque hasta el momento no existe un consenso sobre la estratificación de riesgo de pacientes con SBr, un episodio previo de MSC y el síncope son marcadores de alto riesgo para presentar arritmias ventriculares. La tasa de recurrencia de las arritmias ventriculares en pacientes con MSC abortada y SBr es del 17-62% a los 48-84 meses de seguimiento y la tasa de recurrencia de las arritmias ventriculares en pacientes con síncope y SBr del 19.6% a 24-39 meses seguimiento (Brugada et al, 2002, 2003; Priori et al, 2002; Zipes et al, 2006; Probst et al, 2010; Berne y Brugada et, 2012). Igualmente, los pacientes con síncope recurrente, respiración agónica durante el sueño o convulsiones de causa desconocida en presencia espontánea de un patrón ECG tipo 1 identifica a los sujetos con mayor riesgo de SCD. Sin embargo, cuando el tipo 1 ECG se desencadena a través de un test farmacológico, pero no está presente de forma espontánea, el riesgo arrítmico es menor. Por tanto, el reto es identificar y estratificar mejor el riesgo arrítmico en este grupo de pacientes, para poder seleccionbar aquéllos que deberían recibir un DAI. El valor de la estimulación eléctrica programada es muy debatido y no disponemos de datos prospectivos concluyentes (Priori et al., 2002; Brugada et al. 2002,2003; Probst et al., 2010); una historia familiar de MSC y/o la presencia de una mutación genética no influye en el riesgo arritmogénico. El síndrome de Brugada es extremadamente maligno. En los pacientes que sufren de síncope y en los pacientes recuperados de una casi MSC la incidencia de un nuevo episodio de FV es muy alta. El pronóstico de los pacientes asintomáticos es igualmente malo, ya que hasta un 10% de aquellos pacientes en los el síndrome se diagnosticó en un ECG de rutina presentará un episodio de FV en los dos años siguientes al diagnóstico. La educación y los cambios de estilo de vida para la prevención de las arritmias son críticos en pacientes con SBr (Priori et al., 2015). Se debe enseñar a todos los pacientes con un ECG de Brugada a tratar agresivamente cualquier episodio de fiebre y a evitar fármacos que producen el síndrome (http://www.brugadadrugs.org).
2. Estimulación cardiaca. Debido a que los episodios arrítmicos y la MSC en el SBr ocurren generalmente durante el sueño o en reposo y están asociados con ritmos cardíacos lentos podría existir un papel terapéutico potencial para la implantación de un marcapaso cardíaco (van den Berg et al., 2001). Algunos estudios sugieren que el valor predictivo de los estudios electrofisiológicos puede mejorarse limitando el protocolo de estimulación eléctrica a 2 extrastímulos (Sroubeck et al., 2016), pero esta observación no es apoyada por otros (Priori et al., 2012).
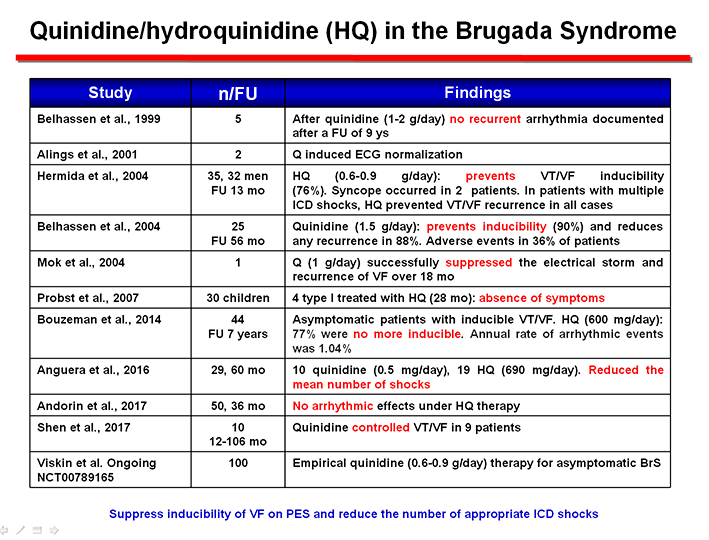
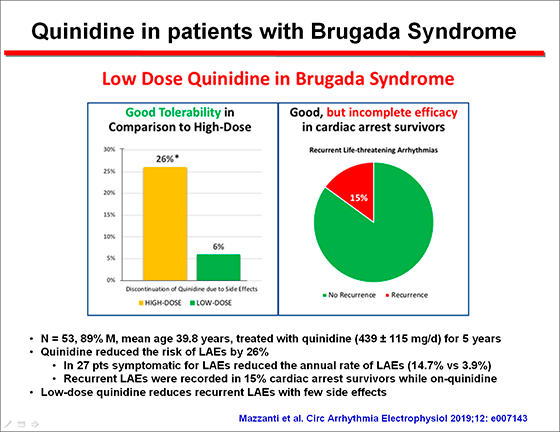
Recientemente, Mazzanti et al (2019) estudiaron 53 pacientes que fueron tratados con quinidina (439 ± 115 mg/dia) durante 5 años, observandpo que a estas bajas dosis la quinidina reducía la incidencia de arritmias que ponían en peligro la vida del paciente en un 26%. Sin embargo, durante el tratamiento con quinidina un 15% de los pacientes desarrollaron este tipo de eventos. Lo interesante es que la discontinuación del tratamiento a estas dosis de quinidina era muy inferior con respecto al uso de dosis plenas de quinidina (6% vs 26%).
La aparición de FV espontáneas en pacientes con SBr se relaciona a menudo con aumentos en el tono vagal y responden a un aumento del tono simpático. De hecho, la infusión i.v. de isoproterenol (0.1 to 1-3 μg/min), que aumenta la corriente de entrada de Ca2+ (ICa) y restaura la morfología de los potenciales de acción epicárdicos y reduce la dispersión transmural de la repolarización, se ha utilizado con éxito para controlar las tormentas arrítmicas y para suprimir la inducción de TV/FV tras estimulación programada, particularmente en niños (Class IIa)(Maury et al., 2004, 2005; Suzuki et al. 2000; Tanaka et al., 2001; Mok et al., 2004; Belhassen et al., 2002; Shimizu et al., 2001; Watanabe et al., 2006; Antzelevitch y Fish, 2006; Ohgo et al., 2007; Pellegrino et al., 2013; Priori et al., 2013).
Ackerman M, Priori S, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Heart Rhythm 2011;8:1308-1339. Agac MT, Erkan H, Korkmaz L. Conversion of Brugada type I to type III and successful control of recurrent ventricular arrhythmia with cilostazol. Arch Cardiovasc Dis 2014;107:476–478. Aizawa Y, Yamakawa H, Takatsuki S, et al. Efficacy and safety of bepridil for prevention of ICD shocks in patients with Brugada syndrome and idiopathic ventricular fibrillation. Int J Cardiol 2013;168:5083–5. Akai J, Makita N, Sakurada H, Set al. A novel SCN5A mutation associated with idio¬pathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Lett. 2000;479:29–34. Amin A, Asghari A, Tan HL. Cardiac sodium channelopathies. Pflugers arch 2010;460:223-237. Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: Report of the Second Consensus Conference: Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659-670. Antzelevitch C, Fish JM. Therapy for the Brugada syndrome. Handb Exp Pharmacol 2006;171:305–330. Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007;115:442–449. Antzelevitch C. Role of transmural dispersion of repolarization in the genesis of drug-induced torsades de pointes. Heart Rhythm. 2005;2(2 Suppl):S9–S15. Antzelevitch C, Patocskai B. Brugada Syndrome: Clinical, Genetic, Molecular, Cellular, and Ionic Aspects. Curr Probl Cardiol. 2016;41:7-57. Antzelevitch C, Yan GX, Ackerman MJ, et al. J-Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. J Arrhythm. 2016;32:315-339. Barajas-Martinez H, Hu D, Ferrer T. Molecular genetic and functional association of Brugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm. 2012;9:548–555. Baroudi G, Pouliot V, Denjoy I, et al. Novel mechanism for Brugada syndrome: defective surface localization of an SCN5A mutant (R1432G). Circ Res 2001;88:E78-83. Behr ER, Savio-Galimberti E, Barc J, et al. Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc Res 2015;106:520–9. Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome.Circulation 2004;110:1731–1737. Belhassen B, Rahkovich M, Michowitz Y, et al. Management of Brugada syndrome: a 33-year experience using electrophysiologically-guided therapy with Class1A antiarrhythmic drugs. Circ Arrhythm Electro- physiol 2015;6:1393–402. Belhassen B, Viskin S, Antzelevitch C. The Brugada syndrome: is an implantable cardioverter defibrillator the only therapeutic option? Pacing Clin Electrophysiol 2002;25:1634–1640. Benito B, Sarkozy A, Mont L, Henkens S, Berruezo A, Tamborero D, et al. Gender differences in clinical manifestations of Brugada syndrome. J Am Coll Cardiol. 2008;52:1567-73. Berne P, Brugada J. Brugada syndrome 2012. Circ J 2012;76:1563-1571. Bezzina C, Veldkamp MW, van den Berg MP, et al. A single Na+ chan¬nel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–1213. Bezzina CR, Barc J, Mizusawa Y. Common variants at SCN5A-SCN10Aand HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013;45:1044–1049. Boczek NJ, Ye D,Johnson EK, et al. Characterization of SEMA3A-encoded semaphorin as a naturally occurring Kv4.3 protein inhibitor and its contribution to Brugada syndrome.CircRes2014;115:460–9. Bouzeman A, Traulle S, Messali A, et al. Long-term follow-up of asymptomatic Brugada patients with inducible ventricular fibrillation under hydroquinidine. Europace 2014;16:572–577. Brugada P, Brugada J, Roy D. Brugada syndrome 1992–2012: 20 years of scientific excitement, and more. Eur Heart J 2013;34:3610–3615. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391-1396. Brugada J, Brugada R, Antzelevitch C, et al. Long-term follow-up of individuals with the electrocardiographic pattern of right bundle-branch block and ST-segment elevation in precordial leads V1 to V3. Circulation. 2002;105:73-78. Brugada J, Brugada R, Brugada P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome and no previous cardiac arrest. Circulation. 2003;108:3092-3096. Brugada P, Brugada R, Brugada J. Patients with an asymptomatic Brugada electrocardiogram should undergo pharmacological and electrophysical testing. Circulation 2005;112:279–285. Brugada J, Brugada R, Brugada P. Pharmacological and device approach to therapy of inherited cardiac diseases associated with cardiac arrhythmias and sudden death. J Electrocardiol. 2000;33(Suppl):41–47. Brugada J, Pappone C, Berruezo A, et al. Brugada syndrome phenotype elimination by epicardial substrate ablation. Circ Arrhythm Electrophysiol. 2015;8:1373–1381. Burashnikov E, Pfeiffer R, Barajas-Martinez H, et al. Mutations in the cardiac L-type calcium channel associ¬ated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm. 2010;7:1872–1882. Cerrone M, Lin X, Zhang M, et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2014;129:1092–103. Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998;392:293–296. Chiale PA, Garro HA, Fernandez PA, et al. High-degree right bundle branch block obscuring the diagnosis of Brugada electrocardiographic pattern. Heart Rhythm 2012;9:974–6. Chinushi M, Aizawa Y, Ogawa Y, et al. Discrepant drug action of disopyramide on ECG abnormalities and induction of ventricular arrhythmias in a patient with Brugada syndrome. J Electrocardiol. 1997;30:133–136. Conte G, Sieira J, Ciconte G, et al. Implantable cardioverter-defibrillator therapy in Brugada syndrome: a 20-year single-center experience. J Am Coll Cardiol 2015;65:879–88. Coronel R, Casini S, Koopmann TT, et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: A combined electrophysiological, genetic, histopathologic, and computational study. Circulation 2005;112:2769-2777. Delpon E, Cordeiro JM, Nunez L, Thomsen PEB, Guerchicoff A, Pollevick GD, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1:209-18. Di Diego JM, Cordeiro JM, et al. Ionic and cellular basis for the predominance of the Brugada syndrome phenotype in males. Circulation 2002;106:2004–2011. DiFrancesco D. Funny channel gene mutations associated with arrhythmias. J Physiol. 2013;591:4117–4124. Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are tem¬perature dependent. Circ Res. 1999;85:803–809. Elizari MV, Levi R, Acunzo RS, et al. Abnormal expression of cardiac neural crest cells in heart development: a different hypothesis for the etiopathogenesis of Brugada syndrome. Heart Rhythm. 2007;4: 359–365. Ezaki K, Nakagawa M, Taniguchi Y, et al. Gender differences in the ST segment: Effect of androgendeprivation therapy and possible role of testosterone. Circ J 2010;74:2448-2454. Frustaci A, Priori SG, Pieroni M, et al. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 2005;112:3680-3687. Fukuyama M, Ohno S, Makiyama T, et al. Novel SCN10A variants associated with Brugada syndrome. Europace 2015;18:905–11. George AL. Inherited disorders of voltage-gated sodium channels. J. Clin. Invest.2005; 115:1990–1999. Giudicessi JR, Ye D, Tester DJ, et al. Transient outward current (Ito) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm 2011;8:1024-1032. Grant AO, Carboni MP, Neplioueva V, et al. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J Clin Invest 2002;110:1201–1209. Hermida JS, Denjoy I, Clerc J, et al. Hydroquinidine therapy in Brugada syndrome. J Am Coll Cardiol 2004;43:1853–1860. Hoogendijk MG, Opthof T, Postema PG, etal. The Brugada ECG pattern: a marker of channelopathy, structural heart disease, or neither?. Toward a unifying mechanism of the Brugada syndrome. Circ Arrhythm Electrophysiol 2010;3:283–90. Hu D, Barajas-Martinez H, Burashnikov E, et al. A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet. 2009;2:270-278. Hu D, Barajas-Martínez H, Medeiros-Domingo A, et al. A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Na(v)1.5 and K(v)4.3 channel currents. Heart Rhythm. 2012;9:760-766. Hu D, Barajas-Martinez H, Pfeiffer R, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol 2014;64:66–79. Huang J-M, Horie M. Genetics of Brugada syndrome. J Arrhythm. 2016; 32: 418–425. Ishikawa T, Sato A, Marcou CA, et al. A novel disease gene for Brugada syndrome: sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hNav1.5. Circ Arrhythm Electrophysiol 2012;5:1098–107. Juang JJ, Horie M. Genetics of Brugada syndrome. J Arrhythm. 2016;32:418-425. Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010;7:33–46. Kattygnarath D, Maugenre S, Neyroud N, et al. MOG1: a new susceptibility gene for Brugada syndrome. Circ Cardiovasc Genet. 2011;4:261-268. Kusano KF, Taniyama M, Nakamura K, et al. Atrial fibrillation in patients with Brugada syndrome relationships of gene mutation, electrophysiology, and clinical backgrounds. J Am Coll Cardiol. 2008;51:1169-75. Kyndt F, Probst V, Potet F, et al. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation. 2001;104:3081–3086. Lambiase PD, Ahmed AK, Ciaccio EJ, Brugada R, Lizotte E, Chaubey S, Ben-Simon R, Chow AW, Lowe MD, McKenna WJ. High-density substrate mapping in Brugada syndrome: combined role of conduction and repolarization heterogeneities in arrhythmogenesis. Circulation 2009; 120:106-117, 101-104. Liu H, Chatel S, Simard C, et al. Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PLoS One 2013;8:e54131. London B, Michale M, Mehdi H, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPDL-1) decreases cardiac Na-current and causes inherited arrhthmias. Circulation. 2007;116:2260-2268. Makita N, Behr E, Shimizu W, et al. The E1784K mutation in SCN5A is associated with mixed clinical phenotype of type 3 long QT syndrome. J Clin Invest. 2008;118:2219–2229. Maury P, Couderc P, Delay M, et al. Electrical storm in Brugada syndrome successfully treated using isoprenaline. Europace 2004;6:130–133. Maury P, Hocini M, Haissaguerre M. Electrical storms in Brugada syn¬drome: review of pharmacologic and ablative therapeutic options. Indian Pacing Electrophysiol J. 2005;5:25–34. Medeiros-Domingo A, Tan BH, Crotti L, et al. Gain-of-function mutation S422L in the KCNJ8-encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm. 2010;7:1466-1471. Meregalli PG, Tan HL, Probst V, et al. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm 2009;6:341-348. Meregalli PG, Wilde AA, Tan HL. Pathophysiological mechanisms of Brugada syndrome: depolarisation disorder, repolarisation disorder or more? Cardiovasc Res. 2005;67:367-378. Mizasuwa Y, Wilde AA. Brugada syndrome. Circ Arrhythm Electrophysiol 2012;5:606-616. Mizusawa Y, Sakurada H, Nishizaki M, et al. Effects of low-dose quinidine on ventricular tachyarrhythmias in patients with Brugada syndrome: low-dose quinidine therapy as an adjunctive treatment. J Cardiovasc Pharmacol. 2006;47:359–364. Mok NS, Chan NY, Chi-Suen CA. Successful use of quinidine in treatmentof electrical storm in Brugada syndrome. Pacing Clin Electrophysiol 2004;27: 821–823. Murakami M, Nakamura K, Kusano KF, et al. Efficacy oflow-dose bepridil for preventionofventricular fibrillation in patients with Brugada syndrome with and without SCN5A mutation. J Cardiovasc Pharmacol 2010;56:389–95. Nademanee K, Veerakul G, Chandanamattha P, et al. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation. 2011;123:1270–1279. Nagase S, Kusano KF, Morita H, et al. Epicardial electrogram of the right ventricular outflow tract in patients with the Brugada syndrome: using the epicardial lead. J Am Coll Cardiol 2002;39:1992-1995. Nielsen MW, Holst AG, Olesen SP, et al. The genetic component of Brugada syndrome. Front Physiol. 2013;4:179. Ohgo T, Okamura H, Noda T, et al. Acute and chronic management in patients with Brugada syndrome associated with electrical storm of ventricular fibrillation. Heart Rhythm 2007;4: 695-700. Ohno S, Zankov DP, Ding WG, et al. KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ Arrhythm Electrophysiol. 2011;4:352-361. Pellegrino PL, Di BM, Brunetti ND. Quinidine for the management of electrical storm in an old patient with Brugada syndrome and syncope. Acta Cardiol 2013;68:201–3. Postema PG, van Dessel PF, de Bakker JM, et al. Slow and discontinuous conduction conspire in Brugada syndrome: a right ventricular mapping and stimulation study. Circ Arrhythm Electrophysiol 2008;1:379-386. Postema PG, van Dessel PF, Kors JA, et al. Local depolarization abnormalities are the dominant pathophysiologic mechanism for type 1 electrocardiogram in brugada syndrome a study of electrocardiograms, vectorcardiograms, and body surface potential maps during ajmaline provocation. J Am Coll Cardiol 2010;55:789-797. Priori S.G., Blomstrom-Lundqvist C., Mazzanti A, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36:2757–2759. Priori SG, Gasparini M, Napolitano C, et al. Risk stratification in Brugada syndrome: results of the PRELUDE (PRogrammed ELectrical stimUlation preDictive valuE) registry. J Am Coll Cardiol 2012;59:37-45. Priori S.G., Wilde A.A., Horie M. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10:1932–1963. Priori SG, Napolitano C, Gasparini M, et al. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation. 2002;105:1342-7. Probst V, Denjoy I, Meregalli PG et al. Clinical aspects and prognosis of Brugada syndrome in children. Circulation 2007;115:2042–2048. Probst V, Veltmann C, Eckardt L,etal. Long-term prognosis of patients diagnosed with Brugada syndrome: results from the FINGER Brugada Syndrome Registry. Circulation 2010;121:635–643. Probst V et al. The psychological impact of implantable cardioverter defibrillator implantation on Brugada syndrome patients. Europace. 2011;13:1034–9. Riuro H, Beltran-Alvarez P, Tarradas A, et al. A missense mutation in the sodium channel beta2 subunit reveals SCN2B as a new candidate gene for Brugada syndrome. Hum Mutat 2013;34:961–6. Sacher F, Jesel L, Jais P, et al. Insight into the mechanism of Brugada syndrome: epicardial substrate and modification during ajmaline testing. Heart Rhythm. 2014;11:732–734. Sacher F, Probst V, Maury P. Outcome after implantation of cardioverter- defibrillator in patients with Brugada syndrome: a multicenter study—part 2. Circulation. 2013;128:1739–1747. Schulze-Bahr E, Eckardt L, Breithardt G, et al. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Hum. Mutat. 2003:21:651–652. Shah AJ, Hocini M, Lamaison D. Regional substrate ablation abolishes Brugada syndrome. J Cardiovasc Electrophysiol. 2011;22:1290–1291. Shimizu W, Kamakura S .Catecholamines in children with congenital long QT syndrome and Brugada syndrome. J Electrocardiol 2001;34:173–5. Shinohara T, Ebata Y, Ayabe R, et al. Combination therapy of cilostazol and bepridil suppresses recurrent ventricular fibrillation related to J-wave syndromes. Heart Rhythm 2014;11:1441–1445. Shirai N, Makita N, Sasaki K, et al. A mutant cardiac sodium channel with multiple biophysical defects associated with overlapping clinical fea¬tures of Brugada syndrome and cardiac conduction disease. Cardiovasc Res. 2002;53:348–354. Smits JP, Eckardt L, Probst V, et al. Genotype-phenotype relationship in Brugada syndrome: electrocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients. J Am Coll Cardiol 2002;40:350–356. Smits JP, Koopmann TT, Wilders R, et al. A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. J Mol Cell Cardiol 2005;38:969-981. Sroubek J, Probst V, Mazzanti A. Programmed Ventricular Stimulation for Risk Stratification in the Brugada Syndrome: A Pooled Analysis. Circulation. 2016;133:622-30. Sugao M, Fujiki A, Nishida K, et al. Repolarization dynamics in patients with idiopathic ventricular fibrillation: pharmacological therapy with bepridil and disopyramide. J Cardiovasc Pharmacol 2005;45:545–549. Sumi S, Maruyama S, Shiga Y, et al. High efficacy of disopyramide in the management of ventricular fibrillation storms in a patient with Brugada syndrome. Pacing Clin Electrophysiol. 2010;33:e53-6. Suzuki H, Torigoe K, Numata O, et al. Infant case with a malignant form of Brugada syndrome. J Cardiovasc Electrophysiol 2000;11:1277–1280. Takehara N, Makita N, Kawabe J, et al. A cardiac sodium channel mutation identified in Brugada syndrome associated with atrial standstill. J Intern Med. 2004;255:137-142. Tanaka H, Kinoshita O, Uchikawa S, et al. Successful prevention of recurrent ventricular fibrillation by intravenous isoproterenol in a patient with Brugada syndrome. Pacing Clin Electrophysiol 2001;24:1293–4. Tanaka H, Kinoshita O, Uchikawa S, et al. Successful prevention of recurrent ventricular fibrillation by intravenous isoproterenol in a patient with Brugada syndrome. Pacing Clin Electrophysiol 2001;1293-1294. Tester DJ, Will ML, Haglund CM, et al. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2005;2:507–517. Tsuchiya T, Ashikaga K, Honda T, et al. Prevention of ventricular fibrillation by cilostazol, an oral phosphodiesterase inhibitor, in a patient with Brugada syndrome. J Cardiovasc Electrophysiol 2002;13:698–701. Tsuchiya T, Ashikaga K, Honda T, et al. Ueda K, Hirano Y, Higashiuesato Y, et al. Role of HCN4 channel in preventing ventricular arrhythmia. J Hum Genet. 2009;54:115-121. Ueda K, Nakamura K, Hayashi T, et al. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J Biol Chem 2004;279:27194-27198. Valdivia CR, Tester DJ, Rok BA, et al. A trafficking defective, Brugada syn¬drome-causing SCN5A mutation rescued by drugs. Cardiovasc Res. 2004;62:53–62. van Den Berg MP, Wilde AA, Viersma TJW. Possible bradycardic mode of death and successful pacemaker treatment in a large family with features of long QT syndrome type 3 and Brugada syndrome. J Cardiovasc Electrophysiol. 2001;12:630–636. Veerakul G, Nademanee K. Brugada syndrome: two decades of progress. Circ J 2012;76:2713-2722. Verkerk AO, Wilders R, Schulze-Bahr E, et al. Role of sequence variations in the human ether-ago-go-related gene (HERG, KCNH2) in the Brugada syndrome 1. Cardiovasc Res 2005;68:441-453. Viskin S, Wilde AA, Tan HL, et al. Empiric quinidine therapy for asymptomatic Brugada syndrome: time for a prospective registry. Heart Rhythm 2009;6:401–4. Wang DW, Makita N, Kitabatake A., et al. Enhanced Na+ channel intermediate inactivation in Brugada syndrome. Circ. Re 2000; 87:E37-E43. Wang Q, Ohno S, Ding WG. Gain-of-function KCNH2 mutations in patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2014;25(5):522–530. Watanabe A, Fukushima KK, Morita H, et al. Low-dose isoproterenol for repetitive ventricular arrhythmia in patients with Brugada syndrome. Eur Heart J 2006;27:1579–83. Watanabe H, Koopmann TT, Le Scouarnec S, et al. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Invest 2008;118: 2260-2268. Weiss R, Barmada MM, Nguyen T, et al. Clinical and molecular heterogeneity in the Brugada syndrome: a novel gene locus on chromosome 3. Circulation. 2002;105:707–713. Wilde AA, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation 2002;106:2514–2519. Wilde AA, Postema PG, Di Diego JM, et al. The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J Mol Cell Cardiol. 2010;49:543–553. Wilde AA, Viskin S. EP testing does not predict cardiac events in Brugada syndrome. Heart Rhythm 2011;8:1598-1600. Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100:1660-1666. Zhang J, Sacher F, Hoffmayer K et al. Cardiac electrophysiological substrate underlying the ECG phenotype and electrogram abnormalities in Brugada syndrome patients. Circulation 2015;131:1950–1959. Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation 2006;114:e385–484. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

|
|
| Aviso legal Esta obra está bajo una licencia de Creative Commons Reconocimiento-NoComercial-SinObraDerivada 4.0 Internacional |
