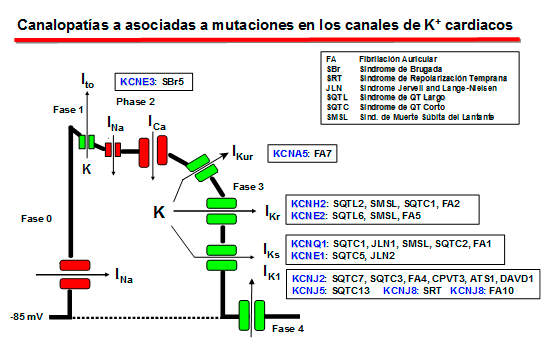
Canales cardiacos de Potasio
|
Papel fisiológico de los canales de K+ Clasificación de los canales de K+ A) Canales de K que presentan 6 segmentos transmembrana y 1 poro (6TM-1P)
Papel fisiológico de los canales de K+ Los canales de K+ constituyen el grupo más numeroso, heterogéneo y ubicuo de proteínas estructurales de membrana. En las células cardiacas, los canales de K+ juegan un importante papel en el mantenimiento del potencial de reposo celular, determinan la forma y duración del potencial de acción, la frecuencia de disparo de las células automáticas, la liberación de neurotransmisores y la morfología y duración de los PA y constituyen la diana sobre la que actúan diversos neurotransmisores, hormonas, fármacos y toxinas que modulan la función cardiaca (Tamargo et al., 2004; Nerbonne y Kass 2005). Clasificación de los canales de K+ Los canales de K+ se pueden clasificar atendiendo a su topología, es decir, al número de poros y de segmentos transmembrana (TM) del canal (denominados S en los canales Kv y M en los canales no activados por voltaje), así como atendiendo a las diferencias en sus propiedades funcionales y farmacológicas. Los canales de K+ están formados por el ensamblaje de subunidades α que forman el poro del canal y diversas subunidades β. Se han identificado y clonado más de 80 genes que codifican diversos canales de K+ humanos (Alexander et al., 2011; Coetzee et al., 1999; Jenkinson et al., 2006; Lüscher y Slesinger 2010; Nerbonne y Kass 2005; Tamargo et al., 2004).
(1-4,11). Los genes que codifican las subunidades α y β de los canales de K+ cardiacos se muestran en la Tabla 1.
Por otro lado, en las células cardiacas los canales de K+ se pueden clasificar en:
A) Canales de K que presentan 6 segmentos transmembrana y 1 poro (6TM-1P) Los canales funcionales resultan del ensamblaje entre 4 subunidades a formadoras del poro del canal y 4 subunidades , formando homo o heterotetrámeros. Cuando el canal es un homotetrámero, las cuatro subunidades a están codificadas por el mismo gen, mientras que los heterotetrámeros están constituidos por productos de distintos genes, pero siempre de la misma subfamilia.
Es la unidad central del canal, ya que contiene el poro conductor, el filtro de selectividad que permite el paso de K+ frente a otros cationes, el sensor de voltaje que controla los mecanismos que regulan la cinética de apertura y cierre del canal y los puntos de unión para los ligandos endógenos y fármacos. Presenta 6 segmentos que atraviesan la membrana (S1-S6) con estructura secundaria de α-hélice conectadas entre sí, de los que 5 son hidrofóbicos (S1-S3, S5, S6) y uno (S4) está cargado positivamente (contiene arginina o lisina cada 3 residuos), extremos carboxi (C-) y amino (N)-terminales intracelulares (Caterall, 2010). (Cotzee et al., 1998; MacKinnon 1991, 2003; Pongs et al., 1992; Tamargo et al., 2004; Yellen et al., 2002; Caterall 2010). Los iones K+ se conducen de manera muy eficiente (107 iones canal-1seg-1) y son altamente selectivos, siendo al menos 10.000 veces más permeantes para el K+ que para el Na+ (Doyle et al., 1998, Sansom et al., 2002). El poro iónico está formado por los segmentos S5 y S6 y la región peptídica que une S4 y S5 (lazo P) y es responsable de la conducción y selectividad iónica del canal. La boca interna del canal está formado por el segmento que une los segmentos S4 y S5 y las regiones citoplasmáticas de los segmentos S5 y S6, mientras que la boca externa la forman la región P y la parte extracelular de los segmentos S5 y S6. La región P consta de 19 aminoácidos y penetra en el interior de la membrana. Los primeros 8 aminoácidos (D431-V438) y los 3 últimos (D447-T449) se localizan en la boca externa del poro, observándose que la sensibilidad al tetraetilamonio cuando se aplica a la superficie extracelular (TEAo) del canal aumenta casi 50 veces cuando la T449 se reemplaza por tirosina, cisteína o fenilalanina, mientras que la mutación de este residuo por lisina, arginina, glutamato o valina reduce la sensibilidad al TEAo y la amplitud de la corriente y suprime la rectificación del canal. La región P contiene también una secuencia muy conservada [(T/S)xxTxGYG] que determina la selectividad del poro para el K+ (MacKinnon 1991; Heginbotham et al., 1994).
Los canales tipo Shaker adoptan 3 estados conformacionales: un estado abierto (O), y dos no conductores, cerrado (C) e inactivo (I) (Zagotta y Aldrich, 1990). Durante la despolarización celular, los canales Kv se activan de forma inmediata con una cinética sigmoidal que sugiere que deben moverse a través de diversos estados no conductores, primero hacia un estado cerrado desde el cual el canal puede abrirse (C*) y, posteriormente, de C* a O: C ↔ C ↔ Cn ↔ C* ↔ O ↔ I. A continuación los canales entran en un estado no conductor (I), lo que conduce a una disminución de la corriente. La activación del canal implica desplazamientos relativamente grandes del segmento S4 que conducen a la apertura del canal por medio del acoplamiento directo entre la región del lazo entre S4-S5 y el extremo C terminal de S6, que con S5 limita la boca intracelular del poro del canal (Yellen, 2002). El segmento C-terminal de S6 contiene una secuencia altamente conservada (PxP o PxG), que es crítica en el acoplamiento del movimiento del sensor de voltaje con la apertura del canal. Luego, muchos canales se inactivan (se vuelven a los estados no conductores), lo que lleva a una disminución de la corriente macroscópica activada (Jiang et al., 2003). La inactivación del canal puede realizarse desde el estado abierto (O → I) o sin que el canal se abra (C* → I), mientras que la deactivación (I ↔ O) tiene lugar cuando la membrana se hiperpolariza. Cuando la membrana se repolariza los canales pasan del estado inactivo al estado cerrado (I → C) desde el que el canal esta listo una vez más para volver a activarse en respuesta a la despolarización de la membrana.
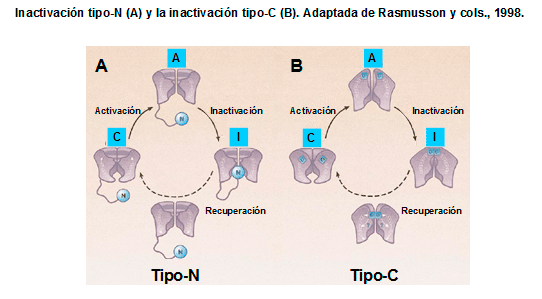
La inactivación lenta tipo-C de los canales Kv1.2, Kv1.5, Kv2.1 y KCNH2 se produce a nivel de la boca externa del poro y parece resultar de cambios conformacionales en el filtro de selectividad del canal (López-Barneo et al., 1993; Rasmusson et al., 1998; Imai et al., 2010; Cuello et al., 2010). Se ha descrito que la alanina-469 localizada en el segmento S6 y la treonina localizada en la porción extracelular del lazo P (Thr449) juegan un papel fundamental en este tipo de inactivación (Hoshi et al., 1990; Pong et al., 1992; López-Barneo et al., 1993). La inactivación tipo-C se suprime cuando se realizan mutaciones en residuos situados en la boca extracelular del canal y en la porción extracelular del segmento S6 y se retrasa tras la aplicación extracelular de tetraetilamonio y al aumentar la [K+]e, debido a la interacción del K+ con un sitio de alta afinidad situado en la cara externa del poro iónico del canal, que impide que se produzca el cierre de la boca del canal, retrasando la inactivación (López-Barneo et al., 1993).
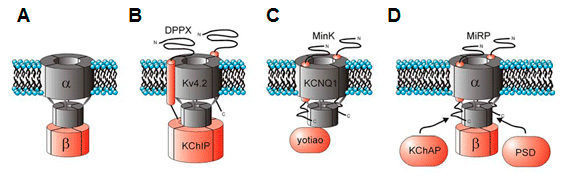
Aunque la expresión de las subunidades α es suficiente para generar canales de K+ funcionales, su coexpresión con subunidades auxiliares o accesorias es necesaria para reproducir las características de los canales nativos (Tabla). Estas subunidades controlan la expresión y/o transporte a la membrana celular de los canales, el gating de los canales y pueden servir como sitio de unión de ligandos exógenos o endógenos que regulan el canal (Hanlon y Wallace, 2002). Subunidad α de los canales de K+ activados por Ca2+(KCa)
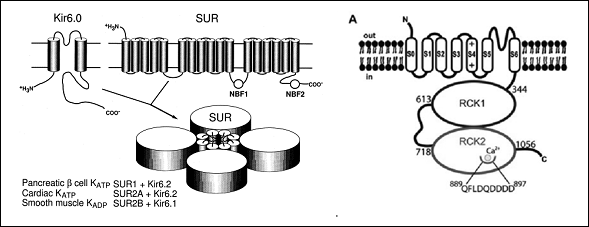
Los monómeros de los canales de K+ de alta conductancia (BKCa) activados por Ca2+(BKCa) están codificados por el gen KCNMA1. La subunidad α presenta (Figura) un extremo N-terminal extracelular corto, 7 segmentos transmembrana (S0-S6). S1-S4 forma el sensor de voltaje (S4 contiene varios residuos de arginina que "detectan" los cambios en la carga y se mueven de manera muy similar a otros canales Kv) y S5-S6 forma el poro del canal y contiene el filtro de selectividad). Estos canales presentan un largo dominio C-terminal intracelular que contiene dos dominios RCK (motifs regulating the conductance of K+), el segundo de los cuales que contienen los puntos de unión para el Ca2+(Yuan et al., 2010) (Figura. Las subunidades β, codificadas por los genes KCNMB1-4, se ensamblan con las subunidades α formando tetrámeros. Las subunidades β 1 y β 2 alteran la dependencia de Ca2+; la β 3 altera el gating del canal (facilitando su inactivación) y la β 4 modula las fosforilación de las subunidades.
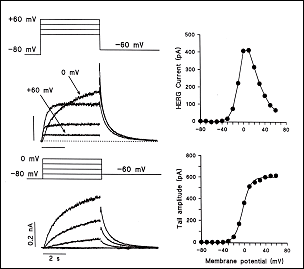
1. El componente de activación rápida de la corriente rectificadora de salida de K+ (IKr) La IKr juega un papel importante en la fase 3 de repolarización cardiaca y en el control de la duración del PA y de la refractariedad auricular y ventricular. La corriente nativa resulta del ensamblaje de la subunidad α (Kv11.1, o HERG) codificada por el gen KCNH2 con dos auxiliares subunidades β, MinK codificada por el gen KCNE1 y MiRP1 codificada por el gen KCNE2) (Sanguinetti, 2010). En humanos, el gen KCNH2 que codifica la subunidad α de los canales de K+ ether a go-go-related type 1 (hERG1) está localizado en el cromosoma 7q35-36, y la región de codificación comprende 16 exones que abarcan aproximadamente 34 kb de secuencia genómica. La subunidad hERG1 está compuesta por 1.159 aminoácidos (127 kDa) y tiene seis dominios transmembrana (S1-S6). hERG1a presenta un largo N-terminal (376 aminoácidos) y los residuos 1-135 contienen una estructura de interacción proteína-proteína llamada dominio Per-Arnt-Sim (PAS) que normalmente interactúa con el canal y reduce la velocidad de desactivación (Morais et al., 1998). El dominio PAS puede ser fosforilado (Cui et al., 2000) y necesita estar plegado correctamente para que tenga lugar el tráfico normal del complejo del canal desde el retículo sarcoplásmico (RS) al aparato de Golgi y, posteriomente, a la superficie celular (Paulussen et al., 2002). La lenta desactivación asociada a las subunidades hERG1a puede estar mediada por una interacción entre el dominio PAS y el lazo de unión entre S4-S5 (Wang et al., 1998), una estructura que acopla el sensor de voltaje al movimiento de apertura y cierre de la puerta de activación (Long et al., 2005). Mutaciones en esta región presentes en pacientes con SQTL interrumpen el tráfico del canal y su deleción acelera en gran medida la velocidad de desactivación (Chen et al, 1999;. Morais et al, 1998) quizás por interrumpir su interacción con el lazo de unión entre los segmentos S4-S5 del canal (Sanguinetti , 2010). El extremo C-terminal del canal contiene un dominio de unión a nucleótidos cíclicos (CNBD). Los canales HERG están regulados por la PKA, que fosforila diversos residuos localizados en los extremos C-y N-terminales, y por el cAMP a nivel del CNBD. La activación de la PKA reduce la corriente hERG, mientras que la unión directa de AMPc al canal hERG la aumenta. El AMPc aumenta la corriente hERG cuando se coexpresa con MiRP1 o MinK, un hallazgo de que mutaciones de pérdida de función en el CNBD se asocian con el SQTL2 (Satler et al, 1996;. Tseng, 2001).
Figura. Corrientes registradas a través de los canles Kv11.1 Canalopatías asociadas a mutaciones en el canal HERG
Mutaciones homocigóticas en el gen KCNH2 son muy raras, ya que conducen a la muerte intrauterina y si los individuos sobreviven presentan una marcada prolongación del intervalo QT grave (Hoorntje et al, 1999; Johnson et al, 2003). Las mutaciones en KCNH2 causantes del SQTL2 pueden resultar en una reducción en IKr por ejercer un efecto dominante negativo (mutaciones localizadas en el poro), por generar canales no funcionales o por una alteración en el tráfico del canal hacia la membrana (Sanguinetti 2010;. Zhou et al, 1998; Splawski y cols ., 2000; Kass y Moss, 2003; Sanguinetti y Tristani-Firouzi, 2006; Sanguinetti 2010). Un plegamiento defectuoso de las proteínas, la retención de proteínas mal plegadas en forma glicosilada en el retículo sarcoplásmico, donde se degradan rápidamente por vía de la de la ubiquitina-proteasoma, o la interrumpieron del tráfico hacia el aparato de Golgi o hacia la superficie de la membrana son los principales mecanismos asociados a la pérdida de función causada por mutaciones con cambio de sentido en hERG1 (Zhou et al 1998, Anderson et al., 2006). Otras mutaciones (p.ej. con desplazamiento del marco de lectura, codon de parada prematura o deleciones que producen una proteína truncada) e incluso algunas mutaciones con cambio de sentido pueden producir una haploinsuficiencia, situación en la que las subunidades mutantes y normales no interactúan y en la que sólo el 50% de los canales hERG es posible que sean disfuncionales o no estén presentes (por haberse degradado rápidamente) (Sanguineti, 2010). 2. El componente lento de la corriente de salida de K+ que presenta rectificación tardía (IKs)
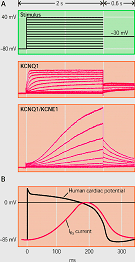
La IKs es la resultante de la coexpresión de la subunidad Kv7.1 (o KVLQT1) codificada por el gen KCNQ1 con una de las cinco subunidades accesorias codificadas por los genes de la familia KCNE (KCNE1-4 - MinK, MiRP1-5) y una proteína adaptadora Yotiao (Barhanin et al., 1996; Sanguinetti y cols, 1996; Marx et al., 2002). El gen KCNQ1 tiene 404 kb de longitud, se encuentra en el cromosoma 11p15.5 y codifica una proteína de 75 kDa que contiene 676 aminoácidos (Wang et al., 1996). Cada subunidad contiene seis segmentos transmembrana (S1-S6 que implican los residuos 122-348) conectados por lazos alternantes intra y extracelulares, un poro (formado por los residuos 300-320) situado entre los segmentos S5 y S6, un extremo N-terminal (residuos 1-121) y un largo C-terminal citosólico (residuos 349-676) que participa en el gating del canal, el ensamblaje de las subunidades y el tráfico de loa canales hacia la membrana (Dvir et al., 2014; Wu et al., 2016). Las cuatro subunidades α se unen a la subunidad MinK (129 aminoácidos con un solo segmento transmembrana) codificada por el gen KCNE1 y la proteína adaptadora Yotiao, y forman un compleo macromolecular que permite permite la conducción de K+ a través del poro hidrofílico ubicado en el centro del complejo. Los segmentos S1-S4 del canal de potasio forman un dominio sensor de voltaje (VSD). La región del poro está compuesta por dos segmentos transmembrana (S5-S6) unidos por un lazo que contiene los aminoácidos conservados que forman el filtro de selectividad (residuos 312–317) y los puntos de unión para diversos bloqueantes de canal y que determina la amplitud de la corriente y la selectividad iónica (Kurokawa et al., 2001; Wu et al., 2016). El extremo C-terminal presenta un motivo de cremallera de leucina (residuos 588–616) a través del cual la proteína de anclaje A-quinasa 9 (AKAP9 o Yotiao) acopla la proteína quinasa A (PKA) y la proteína fosfatasa 1 (PP1) al complejo KVLQT1-KCNE1 (KCNE1-5) (Marx et al., 2002).
La estimulación β-adrenérgica aumenta la fosforilación de la IKs mediada a través de la PKA y produce un marcado aumento de la amplitud de la corriente al aumentar la velocidad de activación y reducir la velocidad de desactivación del canal (Marx et al, 2002; Furokawa et al, 2003). En el extremo C-terminal de KCNQ1 existe un motivo de cierre o cremallera de leucina (“leucine zipper” o ZIP, residuos 588-616) que coordina su unión a la proteína de anclaje de la kinasa A tipo 9 (AKAP9 o yotiao (Clancy y Kass, 2005), que a su vez se une y recluta a la PKA y a la proteína fosfatasa 1 (PP1) al canal. Este complejo, a su vez, regula la fosforilación de la Ser-27 en N-terminal de KCNQ1 (Marx et al., 2002). La fosforilación de la IKs aumenta marcadamente la amplitud de la corriente al acelerar la velocidad de activación del canal (C → O) y reducir la velocidad de deactivación del mismo (O → C), lo que se traduce en una aceleración de la repolarización del PA cardiaco (Furokawa et al., 2003; Marx et al., 2002). Las mutaciones en KCNQ1 o KCNE1 pueden reducir la IKs a través de un efecto dominante negativo, una menor respuesta a la señalización β-adrenérgica (Abbott y Goldstein, 2002; Kurokawa et al., 2003). Las mutaciones en KCNQ1 causan un SQTL por una reducción o una pérdida completa de la IKs, lo que resulta en una reducción de la corriente de repolarización durante el AP (Wang et al., 1996). Estas mutaciones a menudo producen un efecto dominante negativo sobre las subunidades normales, lo que resulta en una reducción >50% de la corriente. En otros casos, hay una pérdida completa de la corriente, que se ha demostrado que resulta del ensamblaje de canales no funcionales o de la imposibilidad de las subunidades sintetizadas para transitar por el citoplasma y alcanzar la membrana (Bianchi et al., 1999). Las mutaciones también pueden alterar las propiedades de activación de canales, que normalmente se manifiestan como una reducción en la velocidad de activación de los canales (R539W, R555C) (Chouabe et al., 1997, 2000), o un aumento en la velocidad de desactivación de canales (S74L, V47F y W87R en KCNE1 y W248R KCNQ1) (Franqueza et al., 1999; Bianchi et al., 1999; Chen et al., 1999; Chouabe et al. 2000; Splawski et al., 2000). La modulación β-adrenérgica de la IKs requiere la unión del AMPc, la proteína quinasa dependiente de AMPc (PKA) y la proteína fosfatasa 1 (PP1) a la subunidad KCNQ1 a través de la proteína yotiao. Yotiao se une a KCNQ1 por un motivo de cremallera de leucina que se interrumpe por una mutación en el C-terminal de KCNQ1 (G589D) y los canales mutantes no responden a la señalización β-adrenérgica. El desacoplamiento del canal de la modulación simpática evita el acortamiento adecuado de la APD en respuesta a los aumentos en la frecuencia cardíaca y aumenta el riesgo de taquiarritmias ventriculares que pueden conducir a la muerte súnbita en en algunos pacientes (Marx et al., 2002).
B) Canales que presentan dos segmentos transmembrana y 1 poro (2 TM-1P)
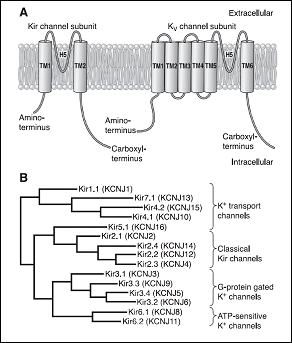
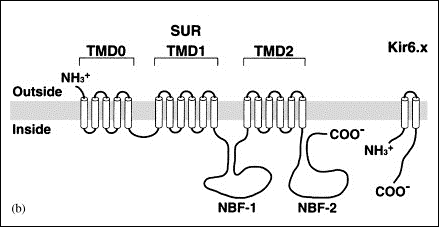
Los canales de K+ rectificadores internos (Kir) codificados por los genes KCNJ1-15 presentan una característica única y es que conducen más iones K+ hacia el interior que hacia el exterior de la célula y conducen iones K+ durante la hiperpolarización, en lugar de durante la despolarización como sucede con otros canales de K+. Por lo tanto, los canales de Kir deben contribuir principalmente a la estabilización del potencial de membrana en reposo. al potencial de equilibrio para K+ (aproximadamente ~ -90 mV) y a la fase de repolarización (fase 3) del PA con poco o ningún efecto sobre la fase de meseta del mismo (Lopatin y Nichols, 2001).
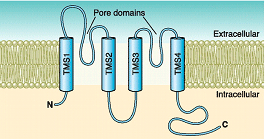
Canalopatías Se han identificado varias canalopatías asociadas con mutaciones por pérdida o ganacia de función en el gen KCNJ2 que codifica loscanales Kir2.1: síndrome de QT largo 7 o síndrome de Andersen-Tawil, taquicardia ventricular polimórfica catecolaminérgica (CPVT), fibrilación auricular familiar y QT3 corto (SQT3). B) Canales que presentan dos segmentos transmembrana y 1 poro (2 TM-1P) Estos canales son muy abundantes tanto en células excitables como no excitables (Enyedi y Czirjak, 2010). Cada canal presenta 4 segmentos transmembrana (TMS1-4) y dos poros y sus extremos N-y C-terminales son intracelulares (Miller et al., 2012; Brohawn et al., 2014; Lolicato et al., 2014; Dong et al., 2016; Bayliss et al., 2019). A diferencia de los canales Kv y Kir que se ensamblan como tetrámeros, los canales K<sub>2P</sub> existen como homodímeros o heterodímeros (Miller et al., 2012; Brohawn et al., 2014; Lolicato et al., 2014; Dong et al., 2016). Cada uno de los (dos) dominios del poro en cada subunidad α contribuye a la formación del poro (Lesage y Lazdunski 2000; Patel y Honore 2001; O'Connel et al, 2002). El motivo G(Y/C)G convencional que confiere la selectividad al K+ se conserva en el lazo del primer poro, pero se sustituye por G(F/L)G en el segundo. Las corrientes generadas por los canales K<sub>2P</sub> muestran dependencia de voltaje y de tiempo y sus curvas I/V puedend escribirse siguiendo la ecuación de Goldman-Hodgkin-Katz (GHK), lo que sugiere que regularían el potencial de membrana en reposo y la excitabilidad celular. Hay cuatro clases de canales K<sub>2P</sub> cardiacos: TASK, TWIK, TREK y THIK.
Abbott GW, Butler MH, Bendahhou S, et al. MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell 2001;104:217-231.
Abbott GW, Goldstein SA. Disease-associated mutations in KCNE potassium channel subunits (MiRPs) reveal promiscuous disruption of multiple currents and conservation of mechanism. FASEB J 2002;16:390-400.
Accili EA, Kiehn J, Yang Q, et al. Separable Kvbeta subunit domains alter expression and gating of potassium channels. J Biol Chem 1997;272:25824-25831.
Alexander SP, Striessnig J, Kelly E, Marrion NV, et al; CGTP Collaborators. (2017) The Concise Guide to PHARMACOLOGY 2017/18: Voltage-gated ion channels. Br J Pharmacol. 174 Suppl 1: S160-S194.
An WF, Bowlby MR, Betty M, et al. Modulation of A-type potassium channels by a family of calcium sensors. Nature 2000;403:553-556.
Anderson CL, Delisle BP, Anson BD, et al. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373.
Anumonwo J, Lopatin A. Cardiac Strong Inward Rectifier Potassium Channels. J Mol Cell Cardiol 2010;48: 45.
Attali B, Chandy G, Hunter M, et al. Voltage-gated potassium channels. Accessed on 30/06/2019. IUPHAR/BPS Guide to PHARMACOLOGY, http://www.guidetopharmacology.org/ GRAC/FamilyDisplayForward?familyId=81.
Bao L, Kefaloyianni E, Lader J, et al. Unique properties of the ATP-sensitive K⁺ channel in the mouse ventricular cardiac conduction system. Circ Arrhythm Electrophysiol 2011;4:926-935.
Barhanin J, Lesage F, Guillemare E, et al. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 1996;384:78-80.
Bayliss DA, Czirják G, Enyedi P, et al. Two P domain potassium channels. Accessed on 30/06/2019. IUPHAR/BPS Guide to PHARMACOLOGY, http://www.guidetopharmacology.org/ GRAC/FamilyDisplayForward?familyId=79.
Bezanilla F. The voltage sensor in voltage-dependent ion channels. Physiol Rev 2000; 80: 555-592.
Bianchi L, Shen ZJ, Dennis AT, et al. Cellular dysfunction of LQT5-minK mutants: abnormalities of I-Ks, I-Kr and trafficking in long QT syndrome. Hum Mol Genet 1999;8:1499-1507.
Billman GE. The cardiac sarcolemmal ATP-sensitive potassium channel as a novel target for anti-arrhythmic therapy. Pharmacol Ther 2008;120:54-70.
Brohawn SG, Campbell EB, MacKinnon R. Physical mechanism for gating and mechanosensitivity of the human TRAAK K+ channel. Nature. 2014;516:126–130.
Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109:30–35.
Cabral JHM, Lee A, Cohen SL, et al. Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell 1998;95:649-656.
Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010;67:915–928.
Chen J, Zou A, Splawski I, et al. Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. J Biol Chem 1999;274:10113–10118.
Chen QY, Zhang DM, Gingell RL, et al. Homozygous deletion in KVLQT1 associated with Jervell and Lange-Nielsen syndrome. Circulation 1999;99: 1344-1347.
Chouabe C, Neyroud N, Guicheney P, et al. Properties of KvLQT1 K_ channel mutations in Romano-Ward and Jervell and Lange-Nielsen inherited cardiac arrhythmias. EMBO J 1997;16:5472-5479.
Chouabe C, Neyroud N, Richard P, et al. Novel mutations in KvLQT1 that affect I-ks activation through interactions with Isk. Cardiovasc Res 2000;45:971-980.
Clancy CE, Kass RS. Inherited and acquired vulnerability to ventricular arrhythmias: Cardiac Na+ and K+ Channels. Ohysiol Rev 2005;85:33-47.
Coetzee W, Amarillo Y, Chiu J, et al. Molecular diversity of K+ channels. Ann N Y Acad Sci 1999;868:233-285.
Cohen A, Ben-Abu Y, Zilberberg N. Gating the pore of potassium leak channels. Eur Biophys J. 2009;39:61-73.
Cuello LG, Jogini V, Cortes DM, et al. Structural mechanism of C-type inactivation in K(?) channels. Nature 2010;466:203–208.
Cui J, Melman Y, Palma E, et al. Cyclic AMP regulates the HERG K+ channel by dual pathways. Curr Biol 2000;10:671–674.
Delpón E, Valenzuela C, Pérez O, et al. Propafenone preferentially blocks the rapidly activating component of delayed rectifier K+ current in guinea-pig ventricular myocytes. Voltage-independent and time-dependent block of the slowly activating component. Circ. Res. 1995;76: 223-235.
Donaldson MR, Jensen JL, Tristani-Firouzi M, et al. PIP2 binding residues of Kir2.1 are common targets of mutations causing Andersen syndrome. Neurology 2003;60:1811-1816.
Dong YY, Pike AC, Mackenzie A, et al. K<sub>2P</sub> channel gating mechanisms revealed by structures of TREK-2 and a complex with Prozac. Science. 2015;347:1256–1259.
Bayliss DA, Czirják g, Enyedi P, et al. Two P domain potassium channels. Accessed on 24/07/2019. IUPHAR/BPS Guide to PHARMACOLOGY, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=79.
Doyle DA, Morais-Cabral J, Pfuetzner RA, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 1998;280:69-74.
Dvir M, Peretz A, Haitin Y. Recent molecular insights from mutated Iks channels in cardiac arrhythmia. Curr Opin Pharmacol. 2014;15:74–82.
England SK, Uebele VN, Shear H, et al. Characterization of a voltage-gated K+ channel beta subunit expressed in human heart. Proc Natl Acad Sci USA 1995;92:6309-6313.
Fedorov VV, Glukhov AV, Ambrosi CM, Kostecki G, Chang R, Janks D, Schuessler RB, Moazami N, Nichols CG, Efimov IR. Effects of KATP channel openers diazoxide and pinacidil in coronary-perfused atria and ventricles from failing and non-failing human hearts. J Mol Cell Cardiol. 2011;51:215–225.
Flagg TP, Kurata HT, Masia R, et al. Differential structure of atrial and ventricular KATP: atrial KATP channels require SUR1. Circ Res 2008;103:1458-65.
Franqueza L, Lin M, Splawski I, et al. Long QT syndrome-associated mutations in the S4–S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J Biol Chem 1999;274:21063-21070.
Grover GJ and Garlid KD. ATP-sensitive potassium channels: a review of their cardioprotective pharmacology. J Mol Cell Cardiol 2000;32:677-695.
Gulbis JM, Zhou M, Mann S, et al. Structure of the cytoplasmic beta subunit-T1 assembly of voltage-dependent K+ channels. Science 2000;289:123-127.
Hanlon MR, Wallace BA. Structure and function of voltage-dependent ion channel regulatory β subunits. Biochemistry 2002;41:2886– 94.
Heginbotham L, Lu Z, Abramson R, MacKinnon R. Mutations in the K+ channel signature sequence. Biophys J 1994;66:1061-1067.
Hibino H, >Inanobe A, Furutani K, et al. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 2010;90:291-366.
Hoorntje T, Alders M, van Tintelen P, et al. Homozygous premature truncation of the HERG protein: the human HERG knockout. Circulation 1999;100:1264–1267.
Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 1990;250:533-538.
Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron 1991;7:547-556.
Huang CL, Feng S, and Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 1998;391:803-806.
Imai S, Osawa M, Takeuchi K, et al. Structural basis underlying the dual gate properties of KcsA. Proc Natl Acad Sci USA 2010;107:6216–6221.
Jenkinson DH. Potassium channels-multiplicity and challenges. Br J Pharmacol 2006; 147(Suppl.1): S63-S71.
Jiang Y, Lee A, Chen J, et al. X-ray structure of a voltage-dependent K+ channel. Nature 2003;423:33–41.
Johnson WH Jr, Yang P, Yang T, et al. Clinical, genetic, and biophysical characterization of a homozygous HERG mutation causing severe neonatal long QT syndrome. Pediatr Res 2003;53:744–748.
Kapplinger JD, Tester DJ, Salisbury BA, et al. Spectrum and prevalence of mutations from the first 2, 500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm 2009;6:1297–1303.
Kass RS, Moss AJ. Long QT sndrome: novel insights into the mechanisms of cardiac arrhythmias. J Clin Invest 2003;112:810-815.
Keating M, Atkinson D, Dunn C. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science. 1991;252:704–706.
Kin Y, Misumi Y, Ikehara Y. Biosynthesis and characterization of the brain-specific membrane protein DPPX, a dipeptidyl peptidase IV-related protein. J Biochem (Tokyo) 2001;129:289-295
Koumi S, Wasserstrom JA. Acetylcholine-sensitive muscarinic K+channels in mammalian ventricular myocytes. Am J Physiol. 1994;266:H1812–H1821
Kuang Q, Purhonen P, Hbert H. Structure of potassium channels. Cell. Mol. Life Sci. 2015;72:3677–3693.
Kurokawa J, Abriel H, Kass RS. Molecular basis of delayed rectifier current I(Ks) in heart. J Mol Cell Cardiol. 2001;33:873–882.
Kurokawa J, Chen L, and Kass RS. Requirement of subunit expression for cAMP-mediated regulation of a heart potassium channel. Proc Natl Acad Sci USA 2003;100: 2122–2127.
Kuryshev YA, Gudz TI, Brown AM, et al. KChAP as a chaperone for specific K+ channels. Am J Physiol Cell Physiol 2000;278:C931-C941.
Lees-Miller JP, Duan Y, Teng GQ, et al. Novel gain-of-function mechanism in K+ channel-related long-QT syndrome: altered gating and selectivity in the HERG1 N629D mutant. Circ Res 2000;86:507-513.
Lesage F and Lazdunski M. Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol 2000;279:F793–F801.
Liman ER, Hess P, Weaver F, et al. Voltage-sensing residues in the S4 region of a mammalian K+ channel. Nature 1991;353:752-756.
Lolicato M, Riegelhaupt PM, Arrigoni C, et al. Transmembrane helix straightening and buckling underlies activation of mechanosensitive and thermosensitive K(2P) channels. Neuron. 2014;84:1198-212.
Lomax AE, Rose RA, Giles WR. Electrophysiological evidence for a gradient of G protein-gated K+ current in adult mouse atria. Br J Pharmacol 2003;140:576–584.
Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science 2005;309:903–908.
Lopatin AN, Nichols CG. Inward rectifiers in the heart: an update on IK1. J Mol Cell Cardiol 2001;33:625–638.
López-Barneo J, Hoshi T, Heinemann SH, et al. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Recept Channels 1993;1:61-71.
Lüscher C, Slesinger PA. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci 2010; 11: 301-315.
MacKinnon R, Yellen G. Mutations affecting TEA blockade and ion permeation in voltage-activated K+ channels. Science 1990;250:276-279.
MacKinnon R. Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature 1991;350:232-235.
MacKinnon R. Potassium channels. FEBS Lett 2003;555:62-65.
Martens JR, Kwak Y-G, Tamkun MM. Modulation of Kv channel α/β subunit interactions. Trends Cardiovasc Med 1999;9:253-258.
Marx SO, Kurokawa J, Reiken S, et al. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 2002;295:496-499.
McCrossan ZA, Abbott GW. The MinK-related peptides. Neuropharmacology 2004;47:787-821.
McDonald TV, Yu Z, Ming Z, et al. A minK-HERG complex regulates the cardiac potassium current I(Kr). Nature 1997;388:289-292.
Miller AN, Long SB. Crystal structure of the human two-pore domain potassium channel K<sub>2P</sub>1. Science. 2012;335:432–436.
Morais Cabral JH, Lee A, Cohen SL, et al. Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell 1998;95:649–655.
Morais-Cabral JH, Zhou Y, MacKinnon R. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature 2001;414:37–42.
Morales MJ, Castellino RC, Crews AL, et al. A novel beta subunit increases rate of inactivation of specific voltage-gated potassium channel alpha subunits. J Biol Chem 1995;270:6272-6277.
Moss AJ, Shimizu W, Wilde .A. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481–2489.
Nadal MS, Ozaita A, Amarillo Y, et al. The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron. 2003;37:449-61.
Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev 2005;85:1205-1253.
O’Connell AD, Morton MJ, Hunter M. Two-pore domain K+ channels-molecular sensors. Biochim Biophys Acta 2002;1566:152 – 61.
Papazian DM, Timpe LC, Jan YN, et al. Alteration of voltage-dependence of Shaker potassium channel by mutations in the S4 sequence. Nature 1991;349:305-310.
Patel AJ, Honore´ E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci 2001;24:339–46.
Patel SP, Campbell DL, Strauss HC. Elucidating KChIP effects on Kv4.3 inactivation and recovery kinetics with a minimal KChIP2 isoform. J Physiol 2002;545:5-11.
Paulussen A, Raes A, Matthijs G, et al. A novel mutation (T65P) in the PAS domain of the human potassium channel HERG results in the long QT syndrome by trafficking deficiency. J Biol Chem 2002;277:48610–48616.
Piechotta PL, Rapedius M, Stansfeld PJ, et al. The pore structure and gating mechanism of K<sub>2P</sub> channels. EMBO J. 2011;30:3607–3619.
Pongs O, Schwarz JR. Ancillary subunits associated with voltage-dependent K+ channels. Physiol Rev. 2010; 90: 755-796.
Pongs O. Molecular biology of voltage-dependent potassium channels. Physiol Rev 1992;72:S69-S88.
Pourrier M, Schram G, Nattel S. Properties, expression and potential roles of cardiac K+ channel accesory subunits: MinK, MiRPs, KChIP and KChAP. J Membr Biol 2003;194:141– 52.
Radicke S, Cotella D, Graf EM, et al. Expression and function of dipeptidyl-aminopeptidase-like protein 6 as a putative beta-subunit of human cardiac transient outward current encoded by Kv4.3. J Physiol. 2005;565:751-756. Rasmusson RL, Morales MJ, Wang S, et al. Inactivation of voltage-gated cardiac K+ channels. Circ Res 1998;82:739-750
Sanguinetti M, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature 2006;440:463-469.
Sanguinetti MC and Jurkiewicz NK. Delayed rectifier outward K_ current is composed of two currents in guinea pig atrial cells. Am J Physiol Heart Circ Physiol 1991;260: H393–H399
Sanguinetti MC, Curran ME, Zou A, et al. Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 1996:384:80-83.
Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J Gen Physiol 1990;96:195-215.
Sanguinetti MC, Xu QP. Mutations of the S4-S linker alter activation properties of HERG potassium channels expressed in Xenopus oocytes. J Physiol 1999;514:667-675.
Sanguinetti MC. HERG1 channelopathies. Pflugers Arch. 2010;460:265-276.
Sansom MS, Shrivastava IH, Bright JN, et al. Potassium channels: structures, models, simulations. Biochim Biophys Acta. 2002;1565:294–307.
Satler CA, Walsh EP, Vesely M, et al. Novel missense mutation in the cyclic nucleotide-binding domain of HERG causes long QT syndrome. Am J Med Genet 1996;65:27-35.
Schram G, Pourrier M, Nattel S. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res 2002;90:939-950.
Seino S, Miki T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol 2003;81:133–76.
Shushi L, Kerem B, Goldmit M, et al. Clinical, genetic, and electrophysiologic characteristics of a new PAS-domain HERG mutation (M124R) causing long QT syndrome. Ann Noninvasive Electrocardiol. 2005;10:334–341.
Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature 1996;379:833-6.
Spector PS, Curran ME, Zou A, et al. Fast inactivation causes rectification of the IKr channel. J Gen Physiol 1996;107:611-619.
Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 2000;102:1178-1185.
Stengl M, Volders PGA, Thomsen MB, et al. Accumulation of slowly activating delayed rectifier potassium current (IKs) in canine ventricular myocytes. J Physiol 2003;551:777–786.
Suh BC, Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys 2008;37:175–195.
Tamargo J, Caballero R, Gómez R, et al. Pharmacology of cardiac potassium channels. Cardiovasc Res 2004;62:9-33.
Tinger A, Aziz Q, Li Y, et al. ATP‐sensitive potassium channels and their physiological and pathophysiological roles. Compr Physiol 2018;8:1463-1511.
Tristani-Firouzi M, Sanguinetti MC. Structural determinants and biophysical properties of HERG and KCNQ1 channel gating. J Mol Cell Cardiol 2003;35:27-35.
Tseng G-N. IKr: the hERG channel. J Mol Cell Cardiol 2001;33:835-849.
Vandenberg CA. Inward rectification of a potassium channel in cardiac ventricular cells depends on internal magnesium ions. Proc Natl Acad Sci USA 1987;84:2560-2564.
Volders P, Stengl M, van Opstal J, et al. Probing the contribution of IKs to canine ventricular repolarization: key role for beta-adrenergic receptor stimulation. Circulation 2003;107:2753-2760.
Wang J, Trudeau MC, Zappia AM, et al. Regulation of deactivation by an amino terminal domain in Human Ether-a-go-go-related Gene potassium channels. J Gen Physiol 1998;112:637–647.
Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 1996;12:17-23.
Wei AD, Gutman GA, Aldrich R, et al., International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacological Reviews. 2005;57: 463–72.
Wible BA, Yang Q, Kuryshev YA, et al. Cloning and expression of a novel K+ channel regulatory protein, KChAP. J Biol Chem 1998;273:11745-11751.
Wu J, Ding WG, Horie M. Molecular pathogenesis of long QT syndrome type 1. J Arrhythm. 2016;32:381-388.
Yellen G. The voltage-gated potassium channels and their relatives. Nature 2002;419:35-42.
Yokoshiki H, Sunagawa M, Seki T, et al. ATP-sensitive K+ channels in pancreatic, cardiac, and vascular smooth muscle cells. Am J Physiol 1998;274:C25-37.
Yu H, Wu J, Potapova I, et al. MinK-related peptide 1: a beta subunit for the HCN ion channel subunit family enhances expression and speeds activation. Circ Res 2001;88:E84-E87.
Yuan P, Leonetti MD, Pico AR, et al. Structure of the human BK channel Ca2+-activation apparatus at 3.0 A resolution. Science 2010;329:182-186.
Zagotta WN, Aldrich RW. Voltage-dependent gating of Shaker A-type potassium channels in Drosophila muscle. J Gen Physiol 1990;95:29-60.
Zeng J, Laurita KR, Rosenbaum DS, et al. Two components of the delayed rectifier K_ current in ventricular myocytes of the guinea pig type. Theoretical formulation and their role in repolarization. Circ Res 1995;77:140–152.
Zhang M, Jiang M, Tseng GN. minK-related peptide 1 associates with Kv4.2 and modulates its gating function: potential role as beta subunit of cardiac transient outward channel? Circ Res 2001;88:1012-1019.
Zhao JT, Hill AP, Varghese A, et al. Not All hERG pore domain mutations have a severe phenotype: G584S has an inactivation gating defect with mild phenotype compared to G572S, which has a dominant negative trafficking defect and a severe phenotype. J Cardiovasc Electrophysiol. 2009;20:923–930.
Zhou Y, Morais-Cabral JH, Kaufman A, et al. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature 2001;414:43–48.
Zhou Z, Gong Q, Epstein ML, January CT. HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. J Biol Chem 1998;273:21061–21066. .
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
| Aviso legal Esta obra está bajo una licencia de Creative Commons Reconocimiento-NoComercial-SinObraDerivada 4.0 Internacional |
