Taquicardia Ventricular Polimórfica Catecolaminérgica (TVPC)
La TVPC es un canalopatía hereditaria causado una alteración en el manejo intracelular de Ca2+ que aparece ya a la edad pediátrica (Leenhardt et al., 1995). Se caracteriza por un patrón de ECG normal en reposo (aunque algunos pacientes presentan bradicardia sinusal y un intervalo QT límite), ausencia de enfermedad cardíaca estructural y aparición de TV bidireccional, síncope y MSC inducidos por la estimulación adrenérgica, especialmente el ejercicio físico o el estrés emocional. Diagnóstico Las manifestaciones clínicas de CPVT usualmente ocurren en la primera década de vida y son provocadas por la actividad física o el estrés emocional (Priori et al., 2002). El diagnóstico se basa principalmente en los antecedentes familiares, los síntomas y la aparición de arritmias durante la prueba de esfuerzo o la administración i.v. de catecolaminas. Durante el ejercicio o la administración de catecolaminas por vía i.v. (en pacientes que no pueden realizar una prueba de ejercicio) los pacientes con TVPC desarrollan extrasístoles ventriculares con frecuencias por encima de 100-120 latidos por minuto, seguidos de un aumento en la complejidad de los extrasístoles ventriculares y rachas cortas de TV no sostenida. Si el ejercicio continua aumenta la duración de los episodios de TV y aparece una TV bidireccional (caracterizada por una rotación de 180º latido a latido del complejo QRS) que confirma el diagnóstico (Napolitano et al., 2007). Algunos pacientes, sin embargo, pueden desarrollar una TV polimórfica durante la prueba de esfuerzo (Cerrone et al., 2009). Finalmente, aparecen FV, síncope y MSC. Una vez que finaliza el ejercicio, los cambios en el ECG se revierten en la misma secuencia en que han aparecido. Algunos pacientes con TVPC también presentan arritmias supraventriculares, principalmente ráfagas de taquicardia supraventricular o fibrilación auricular que se superponen con extrasístoles ventriculares y TV. En los familiares asintomáticos de pacientes con CPVT, la prueba de ejercicio tiene una especificidad del 97% y una sensibilidad del 50% para predecir la presencia de la mutación familiar asociada con el CPVT. El diagnóstico de la TVPC a edades muy jóvenes es un predictor de eventos cardiacos futuros.
El diagnóstico diferencial debe hacerse con el LQT1 oculto y con elsíndromede Andersen-Tawill, ya que estos pacientes pueden presentar síncope o MSC durante el ejercicio (Tester et al., 2005). Una prueba de esfuerzo o de la administración de adrenalina i.v. ayuda al diagnóstico, ya que los pacientes con TVPC presentan una TV bidireccional, mientras que los pacientes con LQT1 oculto muestran una prolongación del intervalo QTc (Ackerman et al., 2002). Los pacientes con síndrome de Andersen-Tawil también pueden presentar una TV bidireccional. Sin embargo, las anormalías asociadas al síndrome de Andersen-Tawil (que afectan a la cabeza, la cara y las extremidades) y la prolongación del intervalo QT ayudan al diagnóstico. Papel de la rianodina y calsecuestrina
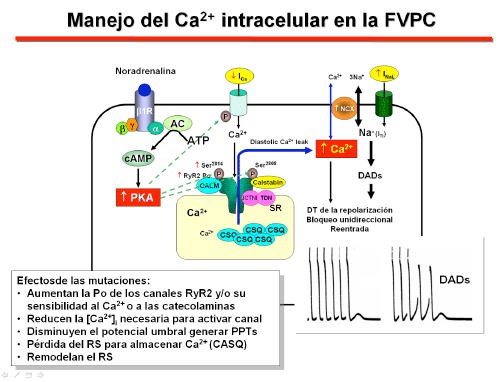
La TVPC es el resultado de una anomalía en la regulación del Ca2+ intracelular, en la que participan dos proteínas localizadas en el retículo sarcoplásmico (RS): el canal de rianodina (RyR2) y la calsecuestrina. El RS es el principal depósito intracelular de Ca2+ y los canales RyR2 controlan la liberación de Ca2+ a partir del RS y desempeña un papel importante en el acoplamiento de excitación-contracción. Este proceso se inicia por la entrada del Ca2+ extracelular tras la activación de los canales de Ca2+ tipo-L durante la fase de meseta del PA cardiaco. Esta entrada de Ca2+ es insuficiente para activar directamente la respuesta contráctil, pero los iones Ca2+ se unen y activan los RyRs, lo que produce la liberación de altas concentraciones de Ca2+ desde el RS hacia el citoplasma y la activación de las proteínas contráctiles. Este mecanismo se conoce como liberación de Ca2+ producida por el Ca2+ (Ca2+-induced Ca2+ release). Durante la diástole, la [Ca2+]i disminuye como consecuencia de la activación de la ATPasa Ca2+-dependiente del RS (SERCA2a), que aumenta la incorporación del Ca2+ intracelular libre en el SR y del intercambiador Na+/Ca2+ (1 Ca2+: 3 Na+) y de la ATPasa Ca2+-dependiente (PMCA) situadas en la membrana celular. Los genes cuyas mutaciones se han asociado a la TVPC se muestran en la Tabla.
Mecanismo arritmogénico de la TVPC.
Implica la activación inducida por las catecolaminas de los receptores β-adrenérgicos que activan la vía de señalización: adenilil-ciclasa - AMPc - activación de la proteína quinasa A dependiente de AMPc cíclico, que fosforila varias proteínas que juegan un papel clave en la regulación del ciclo del Ca2+ , incluyendo el canal RyR2. Ello aumenta la liberación de Ca2+ desde el RS y la [Ca2+]i dutante la diástole . Las mutaciones en los genes RyR2 y CASQ2 aumentan la liberación espontánea de Ca2+ desde el RS durante la diástole. El aumento del Ca2+ intracitoplasmático puede conducir a la activación del intercambiador Na+/Ca2+ que activa una corriente de entrada transitoria despolarizante (ITI), que induce la aparición de pospotenciales tardíos (Figura). Si la amplitud de los pospotenciales tardíos es suficientemente alta como para alcanzar el potencial umbral pueden desencadenar extrasístoles o una TV/FV. Por tanto, los dos pasos más críticos en la arritmogénesis de la TVPC son el aumento en los niveles de catecolaminas y el aumento en la liberación deCa2+ desde el RS durante la diástole, lo que conduce a la aparición de postpotenciales tardíos que desencadenan la TV (Wilde et al., 2008). The risk of arrhythmic events is considered to be higher in patients carrying recessively inherited CASQ2 mutations (the more severe phenotype).
Los mecanismos responsables de la liberación de Ca2+ desde el SR en los canales RyR2 mutados no son bien conocidos. Sin embargo, el potencial arritmogénico de algunas mutaciones es atribuible (Figura): 1) a un marcado aumento en la sensibilidad del canal RyR2 al Ca2+, que resulta en un aumento de la frecuencia de los transientes de Ca2+ que se amplifica por el isoproterenol o al aumentar la frecuencia de estimulación, y disminuye el umbral para la actividad desencadenada (Fernández-Veasco et al., 2009). De hecho, se ha demostrado que la disfunción de los canales RyR2 asociadas a mutaciones en el gen RyR2 aumenta la liberación espontánea deCa2+ y la apaición de pospotenciales tardíos (Kannankeril et al., 2006). 2) Algunas mutaciones sensibilizan el canal a las catecolaminas (es decir, que la liberación de Ca2+inducida tras la activación de la vía AMPc-PKA se produce a niveles de Ca2+ intracitoplasmático más bajos) y/o reducen la [Ca2+] intraluminal umbral necesaria para la activación del canal como consecuencia de una interacción defectuosa entre los dominios del canal RyR2 (Uchinoumi et al., 2010). 3) Una disminución en el umbral para la actividad desencadenada; 4) se produce una pérdida de la capacidad del RS para almecanar Ca2+ pasa al citosol e incrementa la [Ca2+]i. 5) En ocasiones las mutaciones producen un remodelado estructural del RS. Las guías de la ESC de 2015 para el tratamiento de pacientes con arritmias ventriculares y la prevención de muerte súbita cardíaca (Priori et al., 2013) recomiendan:
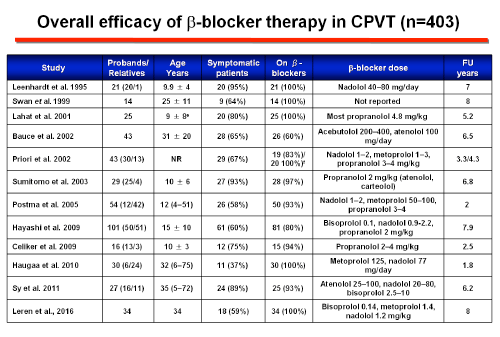
Las guías Europeas (Priori et al., 2015) recomiendan administrar β-bloqueantes en todos los pacientes diagnosticados de TVPC, basado en la presencia de arritmias ventriculares espontáneas o inducidas por el ejercicio; también recomienda administrar β-bloqueantes a familiares con test genético positivo, incluso si tienen un test de ejercicio negativo. Sin embargo, hasta un 30% de los pacientes pueden desarrollar un evento arrítmico (definido como paro cardíaco abortado, descargas apropiadas de DAI y MSC) durante el tratamiento con β-bloqueantes durante un período de seguimiento medio de 8 años (Napolitano y Priori, 2007). Los pacientes que no responden al tratamiento deben considerarse con un riesgo muy alto.
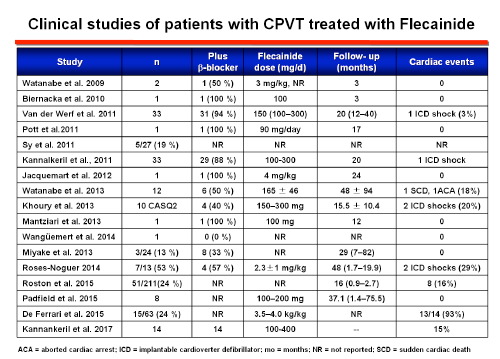
Las guías de la ESC (Priori et al., 2015) le han dado una recomendación de clase IIa al uso de flecainida. El mecanismo por el que la flecainida mejoraría a los pacientes con TVPC incluye: a) un efecto de bloqueo directo en los canales de Na+ dependientes de voltaje que elevaría el umbral para la actividad desencadenada inducida por los postpotenciales tardíos (Bannister et al. 2015; Liu et al., 2011; Sikkel et al., 2013), y b) el bloqueo del estado abierto de los canales RyR2 (o un efecto indirecto sobre los mismos mediado a través de la unión a calmodulina u otros moduladores de RyR2 que disminuye la probabilidad de apertura de los canales, previene la liberación de Ca2+ a través de ldichos canales e inhibe la génesis de las postpotenciales tardíos y el inicio de la TV (Watanabe et al., 2009, 2011). En cardiomiocitos aislados de ratones CASQ2 KO la flecainida reduce la duración de la apertura del canal RyR2 , la frecuencia de los sparks de Ca2+ espontánea y la carga arritmogénia ventricular. Recientemente, se ha demostrado que flecainida disminuye la probabilidad de apertura de canal RyR2 e inhibe la aparición de pospotenciales temprana y de TV (Watanabe et al., 2009).
Un pequeño estudio evaluó la eficacia de la flecainida como monoterapia en 9 pacientes portadores de mutaciones en RYR2, que eran intolerantes o presentaban reacciones adversas a los β-bloqueantes (Padfield GJ et al., 2016). Todos los pacientes respondieron al tratamiento durante una mediana de seguimiento de 37 meses. Sin embargo, aunque la monoterapia con flecainida pudiera considerarse potencialmente una opción en los pacientes que son intolerantes a β-bloqueantes, no se recomienda el uso rutinario de la monoterapia con flecainida porque la evidencia es escasa.
Ackerman MJ, Khositseth A, Tester DJ, et al. Epinephrine–induced QT interval prolongation: a genespecific paradoxical response in congenital long QT syndrome. Clin Proc 2002;77:413–21. Bannister ML, Thomas NL, Sikkel MB, et al. The mechanism of flecainide action in CPVT does not involve a direct effect on RyR2. Circ Res 2015;116:1324-1335. Bhuiyan ZA, Hamdan MA, Shamsi ET, et al. A novel early onset lethal form of catecholaminergic polymorphic ventricular tachycardia maps to chromosome 7p14-p22. J Cardiovasc Electrophysiol 2007;18:1060–1066 Biernacka EK, Hoffman P. Efficacy of flecainide in a patient with catecholaminergic polymorphic ventricular tachycardia. Europace. 2011;13:129-130. Cerrone M, Napolitano C, Priori SG. Catecholaminergic polymorphic ventricular tachycardia: a paradigm to understand mechanisms of arrhythmias associated to impaired Ca2+ regulation. Heart Rhythm 2009;6:1652–1659. Devalla HD, Gélinas R, Aburawi EH, et al. TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol Med. 2016;8:1390-1408. De Ferrari GM, Dusi V, Spazzolini C, et al. Clinical management of catecholaminergic polymorphic ventricular tachycardia: The role of left cardiac sympathetic denervation. Circulation 2015;131:2185-2193. Fernández-Velasco M, Rueda A, Rizzi N, et al. Increased Ca2+ sensitivity of the ryanodine receptor mutant RyR2R4496C underlies catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2009;104:201–9. Ferrero-Miliani L, Holst AG, Pehrson S, et al. Strategy for clinical evaluation and screening of sudden cardiac death relatives. Fundam Clin Pharmacol. 2010;24:619–35. Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J 2004;86:2121–2128 Hayashi M, Denjoy I, Extramiana F, et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation 2009;119: 2426–2434. Hwang HS, Hasdemir C, Laver D, et al. Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol 2011;4:128-35. Jacquemart C, Ould Abderrahmane F, Massin MM. Effects of flecainide therapy on inappropriate shocks and arrhythmias in catecholaminergic polymorphic ventricular tachycardia. J Electrocardiol 2012;45:736-738 Khoury A, Marai I, Suleiman M, et al. Flecainide therapy suppresses exercise-induced ventricular arrhythmias in patients with CASQ2-associated catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2013;10:1671-1675. Laitinen PJ, Brown KM, Piippo K, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001;103:485-90. Leenhardt A, Lucet V, Denjoy I, et al. Catecholaminergic polymorphic ventricular tachycardia in children. A 7–year follow–up of 21 patients. Circulation 1995;91:1512–9. Leenhardt A, Denjoy I, Guicheney P. Catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol 2012;5:1044-1052. Leren IS, Saberniak J, Majid E, et al. Nadolol decreases the incidence and severity of ventricular arrhythmias during exercise stress testing compared with β1-selective β-blockers in patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2016;13:433-440. Liu N, Denegri M, Ruan Y, et al. Flecainide exerts an antiarrhythmic effect in a mouse model of catecholaminergic polymorphic ventricular tachycardia by increasing the threshold for triggered activity [Short communication]. CircRes 2011;109:291-295. Liu N, Rizzi N, Boveri L, Priori SG. Ryanodine receptor and calsequestrin in arrhythmogenesis: what we have learn from genetic diseases and transgenic mice?. J Mol Cell Cardiol 2009;46:149–59. Mantziari L, Vassilikos V, Anastasakis A, et al. A de novo novel cardiac ryanodine mutation (Ser4155Tyr) associated with catecholaminergic polymorphic ventricular tachycardia. Ann Noninvasive Electrocardiol 2013;18:571-576. Maron BJ, Chaitman BR, Ackerman MJ, et al; Working Groups of the American Heart Association Committee on Exercise, Cardiac Rehabilitation, and Prevention; Councils on Clinical Cardiology and Cardiovascular Disease in the Young. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004;109:2807-2816. Miyake CY, Webster G, Czosek RJ, et al. Efficacy of implantable cardioverter defibrillators in young patients with catecholaminergic polymorphic ventricular tachycardia: Success depends on substrate. Circ Arrhythm Electrophysiol 2013;6:579-587. Mohamed U, Gollob MH, Gow RM, et al. Sudden cardiac death despite an implantable cardioverter-defibrillator in a young female with catecholaminergic ventricular tachycardia. Heart Rhythm 2006;3:1486–9. Modi S, Krahn AD. Sudden cardiac arrest without overt heart disease. Circulation. 2011;123:2994–3008. Mohler PJ, Splawski I, Napolitano C, et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci U S A. 2004;101:9137-42. Nyegaard M, Overgaard MT, Søndergaard MT, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91:703–712. Napolitano C, Priori SG. Diagnosis and treatment of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4:675–678. Olde Nordkamp LR, Driessen AH, Odero A, et al. Left cardiac sympathetic denervation in the Netherlands for the treatment of inherited arrhythmia syndromes. Neth Heart J 2014;22:160– Padfield GJ, AlAhmari L, Lieve KV, et al. Flecainide monotherapy is an option for selected patients with catecholaminergic polymorphic ventricular tachycardia intolerant of β-blockade. Heart Rhythm 2016;133:557 – 565. Postma AV, Denjoy I, Kamblock J, et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J Med Genet. 2005;42:863–70. Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001;103:196–200. Priori SG, Wilde AA, Horie M, et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 2013;15:1389–1406. Priori S.G., Blomstrom-Lundqvist C., Mazzanti A, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36:2757–2759. Roses-Noguer F, Jarman JWE, Clague JR, Till J. Outcomes of defibrillator therapy in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2014;11:58-66. Rosso R, Kalman JM, Rogowski O, et al. Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2007;4:1149–54. Roston TM, Vinocur JM, Maginot KR, et al. Catecholaminergic polymorphic ventricular tachycardia in children: Analysis of therapeutic strategies and outcomes from an international multicenter registry. Circ Arrhythm Electrophysiol 2015;8:633-642. Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. 2012;21:2759–2767. Sikkel MB, Collins TP, Rowlands C, et al. Flecainide reduces Ca(2+) spark and wave frequency via inhibition of the sarcolemmal sodium current. Cardiovasc Res 2013;98:286-296. Sumitomo N, Harada K, Nagashima M, et al. Catecholaminergic polymorphic ventricular tachycardia: electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart. 2003;89:66–70. Swan H, Laitinen P, Kontula K, et al. Calcium channel antagonism reduces exercise-induced ventricular arrhythmias in catecholaminergic polymorphic ventricular tachycardia patients with RyR2 mutations. J Cardiovasc Electrophysiol 2005;16:162–6. Swan H, Amarouch MY, Leinonen J, et al. A gain-of-function mutation of the SCN5A gene causes exercise-induced polymorphic ventricular arrhythmias. Circ Cardiovasc Genet. 2014;7:771–781. Tester DJ, Kopplin LJ, Will ML, et al. Spectrum and prevalence of cardiac ryanodine receptor (RyR2) mutations in a cohort of unrelated patients referred explicitly for long QT syndrome genetic testing. Heart Rhythm 2005;2:1099–105. Tiso N, Stephan DA, Nava A, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet 2001; 10:189-94. Uchinoumi H, Yano M, Suetomi T, et al Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ Res. 2010;106:1413–1424. van der Werf C, Kannankeril PJ, et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol 2011;57:2244–54. van der Werf C, Nederend I, Hofman N, et al. Familial evaluation in catecholaminergic polymorphic ventricular tachycardia: disease penetrance and expression in cardiac ryanodine receptor mutation-carrying relatives. Circ Arrhythm Electrophysiol 2012;5:748–756. Watanabe H, Chopra N, Laver D, et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med 2009;15:380–383. Watanabe H, Steele DS, Knollmann BC. Mechanism of antiarrhythmic effects of flecainide in catecholaminergic polymorphic ventricular tachycardia. Circ Res 2011;109:712–713. Watanabe H, van der Werf C, Roses-Noguer F, et al. Effects of flecainide on exercise-induced ventricular arrhythmias and recurrences in genotype-negative patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2013;10:542–7 Wilde AA, Bhuiyan ZA, Crotti L, et al. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. New Engl J Med 2008;358:2024–9. Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation 2006;114:e385–484. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
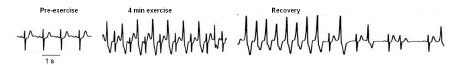
|
|
| Aviso legal Esta obra está bajo una licencia de Creative Commons Reconocimiento-NoComercial-SinObraDerivada 4.0 Internacional |
