|
En los últimos 25 años hemos conocido que existe una correlación entre la presencia de mutaciones en los genes que codifican las subunidades alfa y beta de los canales iónicos o de las proteínas que forman el canalosoma de los canales iónicos cardiacos y la presencia de síndromes arritmogénicos primarios, caracterizados clínicamente por la presencia de taquiarritmias y MSC en pacientes generalmente jóvenes y con un corazón aparentemente normal (Tabla 1). Hablamos, por tanto, de enfermedades de los canales iónicos o canalopatías.
Por tanto, las canalopatías son síndromes arritmogénicos primarios determinados genéticamente, es decir, son enfermedades que alteran las propiedades eléctricas del corazón, pero que no son secundarias a una enfermedad cardíaca estructural subyacente, aunque en alguna canalopatía pueda acompañarse de alguna alteración estructural (p.ej., en el síndrome de Brugada).
Tabla 1. Síndromes arritmogénicos primarios asociados a canalopatías
- Síndrome de QT largo (SQTL) congénito
- Síndrome de QT corto (SQTC)
- Síndrome de Brugada (SBr)
- Síndrome de repolarización temprana (SRT)
- Síndrome del nódulo seno-auricular enfermo (SNS)
- Síndrome de muerte súbita del lactante (SIDS)
- Defectos progresivos de la conducción intracardiaca (DPCI)
- Taquicardias polimórficas ventriculares catecolaminérgicas (TPVC)
- Fibrilación auricular (FA) familiar.
- Fibrilación ventricular (FV) idiotápica
- Parada auricular congénita
- Contracciones prematuras multifocales ectópicas relacionadas con el Purkinje (MEPPC)
- Cardiomiopatía dilatada asociada a arritmias (CMD1E)
- Síndromes que se solapan/superpuestos
Las canalopatías congénitas se asocian a mutaciones en los genes que codifican las subunidades (alfa y auxiliares) de los canales iónicos cardíacos o aquellas que codifican cualquiera de las proteínas que forman el canalosoma de los diferentes canales iónicos cardíacos. Estas mutaciones cambian, a veces solo de manera sutil, el funcionamiento de los canales iónicos que alteran las propiedades eléctricas de los miocitos cardíacos. Por lo tanto, hablamos de CANALOPATÍAS para describir enfermedades causadas por la función alterada de las subunidades de los canales iónicos o las proteínas que las regulan.Las mutaciones pueden aumentar o disminuir la corriente iónica generada por el canal afectado, es decir, que pueden producir una ganacia o una pérdida de la función del canal. La pérdida de funcionalidad conduce con frecuencia a una enfermedad recesiva. Esta reducción de la función puede ser de hasta un 90% en canales tetraméricos (p.ej. de K+). Por otro lado, las mutaciones que producen un aumento de la función del canal se heredan de forma dominante. Este es el caso de las mutaciones de los canales de Na+, que se asocian a la inhibición del proceso de inactivación, a alteraciones en la dependencia de voltaje del proceso de inactivación o a un retraso en el acoplamiento de los procesos de activación e inactivación.
Los canales de K+ son multialélicos, puesto que están formados por proteínas que provienen indistintamente del alelo paterno o el materno. Algunas mutaciones hacen que una o más de esas proteínas que no coensamblen correctamente, de modo que al final hay un menor número de canales en la superficie de la membrana, pero todos los canales situados en la membrana son funcionales. En este caso la mutación produce una haploinsuficiencia, y el porcentaje de canales alterados es ≤ 50%. Sin embargo, en otras ocasiones ocasiones las proteínas mutadas ejercen un efecto dominante negativo, de modo que basta la presencia de una única subunidad mutada para que el canal no sea funcional. En este caso los canales alterados son más del 50%. Por tanto, las mutaciones que producen un efecto dominante negativo confieren una mayor gravedad al cuadro clínico.
Importancia del estudio de las canalopatías
El análisis funcional de las canalopatías nos permite:
-
Conocer mejor los mecanismos implicados en la fisiopatología de los síndromes arritmogénicos primarios y profundizar en los mecanismos implicados en la MSC.
- Conocer el papel funcional de las distintas proteínas que forman parte de los canalosomas de Na+, Ca2+ y K+ en condiciones fisiopatológicas (cómo la mutación altera la función del canal) y analizar la relación existente entre la topología y la función de las distintas subunidades que forman la estructura de los canales iónicos.
- Conocer los determinantes moleculares de las propiedades biofísicas de los canales (p.ej. la selectividad o las características voltaje- y tiempo-dependientes de la cinética de activación, inactivación, y reactivación).
- Estratificar el riesgo del paciente en base a la localización de la mutación. Así, los pacientes con mutaciones localizadas en los segmentos transmembrana o en el poro de la subunidad α de los canales Kv11.1 y Nav1.5 y en los segmentos S5-S6 del canal Kv11.1 ejercen un efecto dominante negativo y se asocian a más eventos cardiacos (síncope y MSC) que las que se localizan en el extremo C-terminal.
- Correlacionar genotipo y fenotipo (Figura).
- Diseñar tests de diagnóstico genético mucho más sensibles y específicas que las previamente utilizadas y nuevas estrategias terapéuticas específicas, “a la carta”, basadas en las consecuencias (aumento o disminución de función) de la mutación responsable de la enfermedad. Así, los bloqueantes de los canales de Na (mexiletina, flecainida) pueden corregir la prolongación del QTc en pacientes con un SQT3 que presentan mutaciones en el gen SCN5A que codifica la subunudad α de los canales de Na+; la normalización de la kalemia (la hipokalemia inhibe la IKr) y algunos agonistas de canales de potasio (nicorandil) corrigen la prolongación del QT en pacientes con un SQT2 asociado mutaciones en el gen KCNH2; por último, la quinidina bloquea la Ito puede normalizar el segmento ST en pacientes con SBr. También se están diseñando estrategias para recuperar el tráfico de los canales mutados hacia la membrana celular.
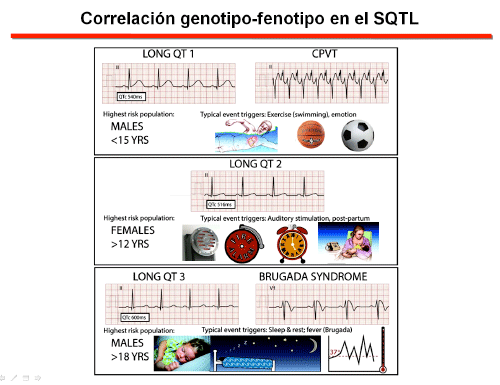
Figura. Correlación genotipo-fenotipo. Algunas canalopatías aparecen durante el ejercicio (SQTL1 y TVPC), durante el sueño o en reposo (SBr y SQTL3) o son inducas por estímulos auditivos o en el postparto (SQTL2). El reconocimiento de aquellos factores que desencadanan la arritmia es de gran importancia para confirmar el diagnóstico. Taken from Skinner et al., 2019.
Referencias
Skinner JR, Winbo A, Abrams D, et al. Channelopathies That Lead to Sudden Cardiac Death: Clinical and Genetic Aspects. Heart Lung Circ. 2019;28:22-30.
|
